Die Summe aller kodierenden Exons des menschlichen Genoms wird als Exom bezeichnet. Obwohl Exons nur 1 %-2 % des Genoms ausmachen, befinden sich circa 89 % aller bekannten krankheitsverursachenden Mutationen in diesen Regionen. Daher ist es zur Klärung von vielen Fragestellungen sinnvoll, eine Exomanalyse durchzuführen.
Die Sequenzierung des gesamten Exoms (“whole exome sequencing”, WES) kann für verschiedene Anwendungsbereiche und Ziele eingesetzt werden, z. B:
- Populationsgenetik
- Genetische Störungen
- Seltene Erkrankungen
- Tumorforschung
Unsere Whole-Exome-Sequencing-Produkte bieten umfassende Lösungen für verschiedene Forschungsfragen.
Unser Service deckt den gesamten Ablauf des Projekts ab: von der Beratung durch erfahrene Experten über eine umfangreiche bioinformatische Auswertung bis hin zu einem übersichtlichen und strukturierten Projektbericht. Der Projektbericht liefert Informationen zur Probenqualität, Sequenzierparametern, bioinformatischen Analysen und Ergebnissen.


CeGaT ist der beste Partner für Ihr Sequenzierprojekt
Unser Produktportfolio für Exome Sequencing
Wir bieten verschiedene Whole-Exome-Sequencing (WES)-Produkte für eine Vielzahl von Forschungsfragen an. Wünschen Sie zusätzlich zu den beinhalteten Leistungen bioinformatische Analysen Ihrer Daten? Jedes unserer Produkte kann durch weitere Dienstleistungen ergänzt werden. Wir beraten Sie gerne.
WES Basic | WES Classic | WES Premium | WES Premium Deep | WES Flex |
Spezies | Spezies | Spezies | Spezies | Spezies Mensch, Maus |
DNA-Qualität | DNA-Qualität | DNA-Qualität | DNA-Qualität | DNA-Qualität |
Anreicherungsmethode | Anreicherungsmethode | Anreicherungsmethode | Anreicherungsmethode | Anreicherungsmethode |
Zielregion | Zielregion | Zielregion | Zielregion | Zielregion |
Sequenzierplattform | Sequenzierplattform | Sequenzierplattform | Sequenzierplattform | Sequenzierplattform |
Output ca. 50-fache Coverage (hängt von der Qualität des Ausgangsmaterials ab) | Output ca. 100-fache Coverage (hängt von der Qualität des Ausgangsmaterials ab) | Output ca. 100-fache Coverage (hängt von der Qualität des Ausgangsmaterials ab) | Output ca. 150-fache Coverage (hängt von der Qualität des Ausgangsmaterials ab) | Output |
Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen |
Unsere Exome-Sequencing-Produkte können mit unseren HLA-Sequencing-Produkten kombiniert werden. Möchten Sie Tumorproben sequenzieren, könnte unser Comprehensive-Tumor-Profiling-Produktportfolio interessant für Sie sein.
Bioinformatik
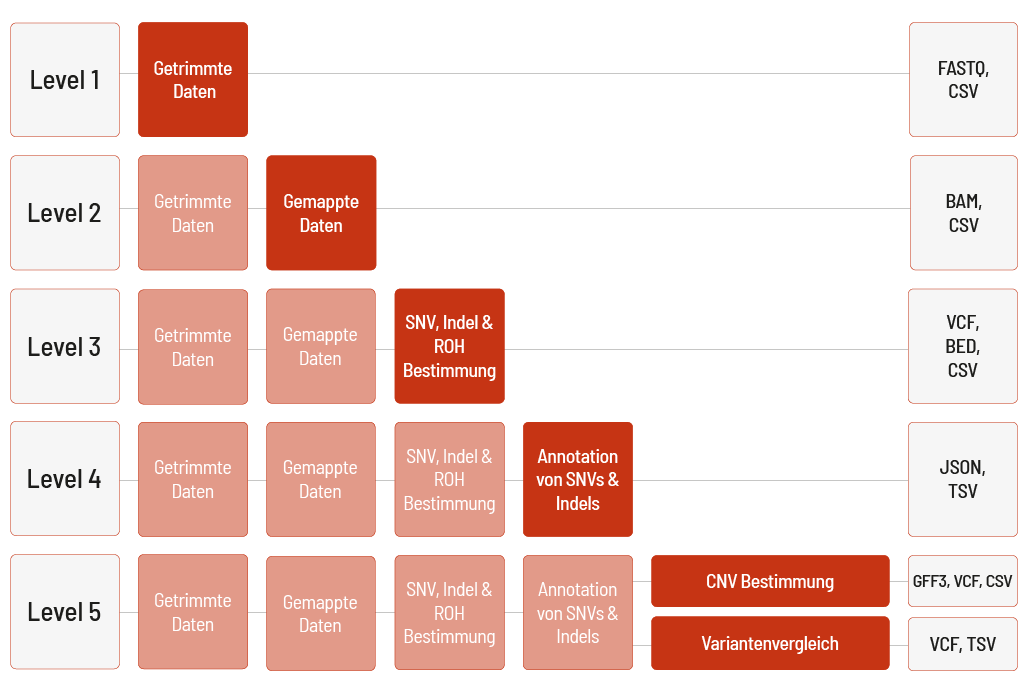
Die Rohdaten der Sequenzierung werden automatisch verarbeitet. Wir bieten verschiedene Level bioinformatischer Analysen an. Das Standardlevel ist Level 1. Mit steigendem Bioinformatiklevel werden mehr Daten geliefert. Alle höheren Level beinhalten dabei die Daten der vorherigen Level. Zusätzlich zu den Daten und unabhängig vom Analyselevel wird ein Projektbericht verfasst.
Alle WES-Produkte werden mit der Illumina DRAGEN Bio-IT-Plattform analysiert. Wir können die Analysen für unsere Standard-WES-Produkte (außer WES Flex) basierend auf den menschlichen Referenzgenomen hg19 oder GRCh38 anbieten.
Level 1:
- Demultiplexing und Adapter-Trimming der Sequenzierdaten (FASTQ-Datei)
- Metriken (CSV-Datei)
- MultiQC-Bericht (HTML-Datei)
Level 2:
- Mapping der Sequenzierdaten (BAM-Datei)
- Metriken (CSV-Datei)
Level 3:
- Bestimmung von Einzelnukleotid-Varianten („single nucleotide variants“, SNVs) und kleinen Insertionen und Deletionen (Indels) (VCF-Datei)
- Bestimmung homozygoter Regionen (ROH) (BED- und CSV-Format)
- Metriken (CSV-Datei)
Level 4:
- Annotation von SNVs und Indels (JSON- und TSV-Datei) (nur für menschliche Proben)
Level 5 (eine der folgenden Möglichkeiten):
- Bestimmung von Kopienzahlveränderungen („copy number variations“, CNVs) (VCF- und GFF3-Datei) inklusive Metriken (CSV-Datei) (nur für menschliche Proben)
- Variantenvergleich mehrerer Proben, z. B. Triofilterung (VCF- und TSV-Datei)
Technische Information
Bei CeGaT wird die Paired-End-Sequenzierung (2 x 100 bp) mit den Sequenzierplattformen von Illumina durchgeführt. Wenn Sie andere Sequenzierparameter benötigen, lassen Sie es uns gerne wissen! Wir können Ihnen weitere Lösungen anbieten.
Weitere Informationen zu Exome Sequencing
Die Exom-Sequenzierung wird auch als Whole Exome Sequencing (WES) bezeichnet. Bei dieser Methode werden alle proteinkodierenden Regionen in einem Genom sequenziert und so genetische Varianten in einer Probe identifiziert. Eine Variation in einer proteinkodierenden Region kann Krankheiten wie die multiple endokrine Neoplasie Typ 2B, Chorea Huntington oder die Creutzfeldt-Jakob-Krankheit verursachen. Neben der Identifizierung solcher krankheitsverursachender Varianten wird Whole Exome Sequencing auch zur Beantwortung von Forschungsfragen eingesetzt. Zu diesen Forschungsfragen zählen die Analyse seltener Varianten, die mit komplexen Merkmalen verbunden sind, oder die Weiterentwicklung der personalisierten Medizin. Darüber hinaus wird Whole Exome Sequencing angewandt, um den Erfolg einer Therapie zu kontrollieren. Whole Exome Sequencing ist also ein wichtiges Instrument für die moderne Medizin sowie für Forschungsfragen, klinische Studien und die Entwicklung von Arzneimitteln.
Die exonischen Regionen beinhalten die Mehrzahl der genetischen Veränderungen, die zu Krankheitsphänotypen führen, darunter große genetische Varianten, Kopienzahlveränderungen (“copy number variations”, CNVs), kleine Insertionen und Deletionen (Indels), Einzelnukleotid-Polymorphismen (“single nucleotid polymorphisms”, SNPs) und Einzelnukleotid-Varianten (“single nucleotid variants”, SNVs).
Folglich wählen Forschende mit Whole Exome Sequencing eine hocheffiziente Methode zur Beantwortung ihrer Forschungsfragen. Darüber hinaus ist Whole Exome Sequencing eine kostengünstige Lösung, um genetische Variationen zu untersuchen, die mit einer bestimmten Krankheit in Verbindung stehen.
Die Technik hinter Whole Exome Sequencing besteht aus zwei Schritten. Im ersten Schritt werden die Regionen erfasst, die für Proteine kodieren, also die Exons. Die erfassten Targets werden isoliert, gewaschen und eluiert. Nach der Amplifikation werden diese Targets im zweiten Schritt verwendet – die Sequenzierung der exomischen DNA.
Whole Exome Sequencing hat im Vergleich zu anderen Technologien, die zur Identifizierung genetischer Varianten angewandt werden können, einige Vorteile. Im Gegensatz zur Microarray-basierten Genotypisierung können mit Whole Exome Sequencing unerwartete genetische Veränderungen identifiziert werden. Die bisherigen Einschränkungen der Mikroarray-basierten Genotypisierung können also mit Whole Exome Sequencing überwunden werden. Im Vergleich zu Whole Genome Sequencing zeigt Whole Exome Sequencing den großen Unterschied zwischen dem Anteil der exonischen und intronischen Regionen: Bei der Exom-Sequenzierung werden nur 1 %-2 % des gesamten Genoms sequenziert. Dies ermöglicht eine höhere Sequenziertiefe und senkt gleichzeitig die Sequenzierkosten.
Unser Engagement für Sie
Schnelle Bearbeitung
Bearbeitungszeit
≤ 15 Werktage
Hohe Qualität
Höchste Genauigkeit bei allen Prozessen
Sichere Datenlieferung
Sichere Bereitstellung der sequenzierten Daten über hausinterne Server
Sichere Aufbewahrung
Sichere Proben- und Datenaufbewahrung nach Projektabschluss
Downloads
Kontaktieren Sie uns
Sie haben noch Fragen oder Interesse an unserem Service? Treten Sie gern mit uns in Kontakt. Wir werden uns schnellstmöglich um Ihr Anliegen kümmern.
Starten Sie Ihr Projekt mit uns
Gerne beraten wir Sie zu unseren Sequenzierdienstleitungen und erarbeiten mit Ihnen gemeinsam die beste Lösung, die auf Ihre klinische Studie oder Forschungsprojekt abgestimmt ist.
Bitte geben Sie, falls möglich, folgende Probeninformationen an: Ausgangsmaterial, Anzahl der Proben, bevorzugte Option für die Vorbereitung der Library, bevorzugte Sequenziertiefe und gewünschte bioinformatische Analysestufe.
