Das Genom bildet die gesamte genetische Information eines Organismus ab. Mit Hilfe der Next-Generation-Sequencing (NGS)-Technologie kann diese vollständig erfasst werden. Durch den Vergleich mit einem Referenzgenom können Veränderungen wie Einzelnukleotid-Varianten (“single nucleotide variants”, SNVs), kleine Insertionen und Deletionen (kurz Indels), Kopienzahlveränderungen (“copy number variations”, CNVs) und Strukturvarianten (“structural variants”, SVs) untersucht werden.
Die Sequenzierung des gesamten Genoms (“whole genome sequencing”, WGS) hilft bei:
- Krebsstudien, Ansätzen der personalisierten Medizin und translationaler Forschung
- der Entdeckung von Biomarkern und dem Verständnis der Pharmakogenetik
- der Erforschung von Krankheiten
- Pflanzen- und Tierzuchtprogrammen
- der Untersuchung von Mikroorganismen
Um eine ausführliche Genomanalyse für Ihr Forschungsprojekt zu erhalten, können Sie zwischen verschiedenen Whole-Genome-Sequencing-Produkten wählen.
Unser Service deckt den gesamten Ablauf des Projekts ab: von der Beratung durch erfahrene Experten über eine umfangreiche bioinformatische Auswertung bis hin zu einem übersichtlichen und strukturierten Projektbericht. Der Projektbericht liefert Informationen zur Probenqualität, Sequenzierparametern, bioinformatischen Analysen und Ergebnissen.


CeGaT ist der beste Partner für Ihr Sequenzierprojekt
Unser Produktportfolio für Genome Sequencing
Wir bieten verschiedene Whole-Genome-Sequencing (WGS)-Produkte für eine Vielzahl von Forschungsfragen an. Unsere WGS-Produkte werden entweder mittels Short-Read-Sequenzierung auf unseren Illumina-Sequenzierplattformen oder mittels Long-Read-Sequenzierung auf unseren PacBio-Sequenzierplattformen prozessiert. Wünschen Sie zusätzlich zu den beinhalteten Leistungen bioinformatische Analysen Ihrer Daten? Jedes unserer Produkte kann durch weitere Dienstleistungen ergänzt werden. Wir beraten Sie gerne.
Short-Read WGS
WGS Large Classic | WGS Small Classic | WGS Flex |
Spezies | Spezies | Spezies |
DNA-Qualität Hochmolekulare DNA | DNA-Qualität Hochmolekulare DNA | DNA-Qualität |
PCR-Amplifikation Ja / Nein | PCR-Amplifikation Ja | PCR-Amplifikation Ja / Nein |
Read-Länge 2 x 150 bp | Read-Länge 2 x 100 bp | Read-Länge 2 x 150 bp oder 2 x 100 bp |
Sequenzierplattform Illumina | Sequenzierplattform Illumina | Sequenzierplattform Illumina |
Output 90 Gb | Output 2 Gb | Output Flexibel |
Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen |
Long-Read WGS
HiFi WGS Classic | HiFi WGS Premium | HiFi WGS Flex |
Spezies Mensch | Spezies Mensch | Spezies Verschiedene |
DNA-Qualität Humane hochmolekulare DNA | DNA-Qualität Humane hochmolekulare DNA | DNA-Qualität Hochmolekulare DNA |
PCR-Amplifikation Nein | PCR-Amplifikation Nein | PCR-Amplifikation Nein |
Read-Länge Im Mittel 15 – 20 kb | Read-Länge Im Mittel 15 – 20 kb | Read-Länge Im Mittel 15 – 20 kb |
Sequenzierplattform PacBio | Sequenzierplattform PacBio | Sequenzierplattform PacBio |
Output 3 – 4 Millionen HiFi-Reads | Output 6 – 8 Millionen HiFi-Reads | Output Projektabhängig |
Beinhaltete Leistungen Projektbericht & Dateien im FASTQ-Format | Beinhaltete Leistungen Projektbericht & Dateien im FASTQ-Format | Beinhaltete Leistungen Projektbericht & Dateien im FASTQ-Format |
Bioinformatik
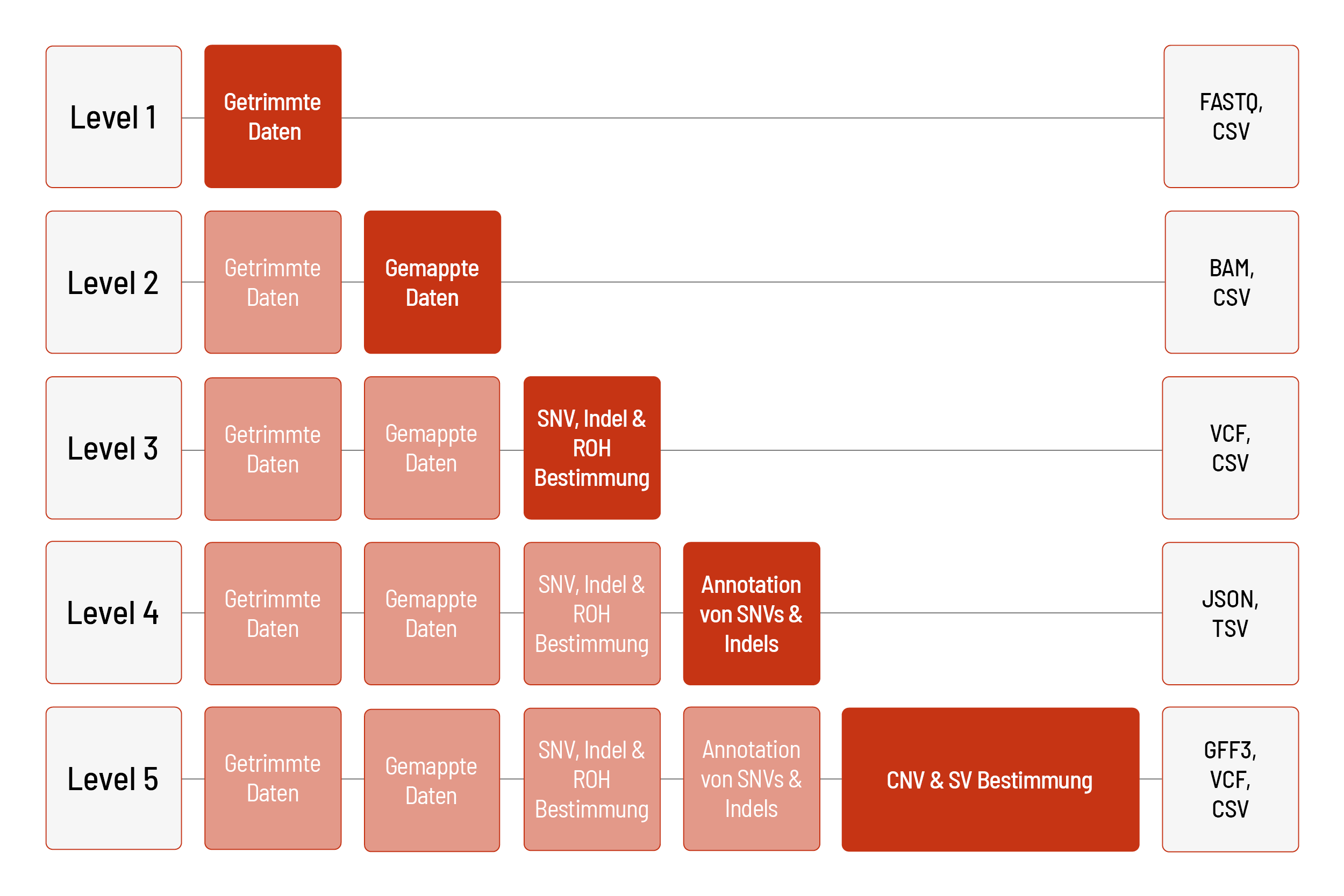
Die Rohdaten der Sequenzierung werden automatisch verarbeitet. Wir bieten verschiedene Level bioinformatischer Analysen an. Das Standardlevel ist Level 1. Mit steigendem Bioinformatiklevel werden mehr Daten geliefert. Alle höheren Level beinhalten dabei die Daten der vorherigen Level. Zusätzlich zu den Daten und unabhängig vom Analyselevel wird ein Projektbericht verfasst.
Analyse der Short-Read-WGS-Produkte
- WGS Large Classic
- WGS Small Classic
- WGS Flex
WGS Large Classic und WGS Flex werden mit der Plattform DRAGEN Bio-IT von Illumina analysiert. WGS Small Classic wird mit der CeGaT-Pipeline analysiert.
Wir können die Analysen basierend auf den menschlichen Referenzgenomen hg19 oder GRCh38 anbieten.
Analyse der Long-Read-WGS-Produkte
- HiFi WGS Classic
- HiFi WGS Premium
- HiFi WGS Flex
Level 3 ist nur für menschliche Proben mit GRCh38 als Referenzgenom möglich.
Level 1:
- Demultiplexing und Adapter-Trimming der Sequenzierdaten (FASTQ-Datei)
- Metriken (CSV-Datei)
- MultiQC-Bericht (HTML-Datei)
Level 2:
- Mapping der Sequenzierdaten (BAM-Datei)
- Vorhersage der Ploidie der Geschlechtschromosomen (CSV-Datei) (nur für menschliche Proben)
- Metriken (CSV-Datei)
Level 3:
- Bestimmung von Einzelnukleotid-Varianten (“single nucleotide variants”, SNVs) und kleinen Insertionen und Deletionen (Indels) (VCF-Datei)
- Ermittlung homozygoter Mutationen (“region of homozygosity”, ROH) (BED- und CSV-Datei) (nur für menschliche Proben)
- Metriken (CSV-Datei)
Level 4:
- Annotation von SNVs und Indels (JSON- und TSV-Datei) (nur für menschliche Proben)
Level 5:
- Bestimmung von Kopienzahlveränderungen (“copy number variations”, CNVs) (VCF- und GFF3-Datei), inklusive Annotationen (JSON- und TSV-Datei) (Annotationen nur für menschliche Proben)
- Bestimmung von Strukturvarianten (“structural variants”, SVs), wie Translokationen, Inversionen sowie große und mittelgroße Indels (VCF- und CSV-Datei), inklusive Annotation (JSON- und TSV-Datei) (Annotationen nur für menschliche Proben)
- Metriken (CSV-Datei)
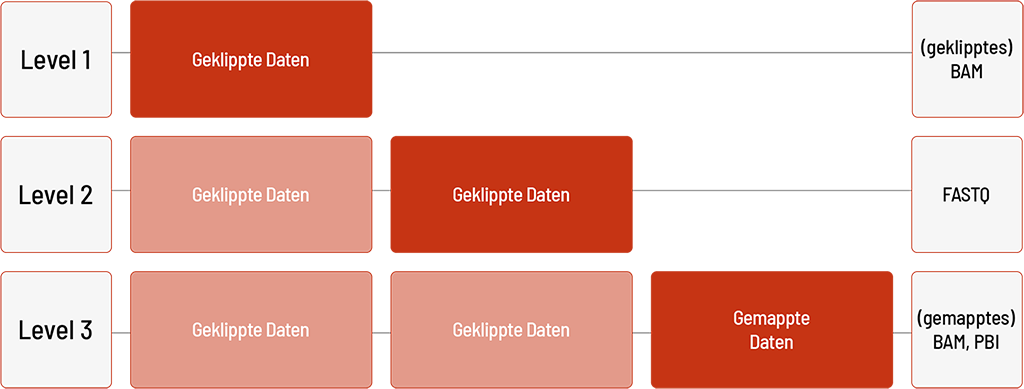
Level 1:
- Geklippte Sequenzierdaten im FASTQ-Format
- Falls erforderlich können geklippte Sequenzierdaten im BAM-Format zusätzlich bereitgestellt werden
Level 2:
- Mapping der Sequenzierdaten (BAM- und PBI-Datei)
Level 3:
- Bestimmung von Einzelnukleotid-Varianten (“single nucleotide variants”, SNVs) und kleinen Insertionen und Deletionen (Indels) (VCF-Datei)
- Bestimmung von Strukturvarianten (“structural variants”, SVs) inklusive Kopienzahlveränderungen („copy number variations“, CNVs) (VCF-Datei)
- Annotation der Varianten (JSON- und TSV-Datei)
- Phasenbestimmung der Varianten (VCF- und BAM-Datei)
- Repeat-Expansions-Analyse (VCF- und BAM-Datei)
Auf Wunsch kann zusätzlich die Bestimmung der Methylierung für menschliche Proben durchgeführt werden. Da die Auswertemöglichkeiten für HiFi WGS Flex stark vom untersuchten Organismus abhängen, kontaktieren Sie uns bitte, damit wir Ihre Optionen besprechen können. Nehmen Sie gerne auch Kontakt mit uns auf, wenn Sie Interesse an einem de-novo-Assembly Ihrer Probe haben.
Technische Informationen
Bei CeGaT wird die Paired-End-Sequenzierung mit den Sequenzierplattformen von Illumina und Pacific Biosciences (PacBio) durchgeführt. Für die Produkte, bei denen die Illumina-Sequenzierplattformen verwendet werden, werden die WGS-Proben entweder im 2 x 150 bp oder 2 x 100 bp Modus sequenziert. Unsere PacBio-Sequenzierplattform ermöglicht uns die Sequenzierung von Fragmenten mit einer mittleren Länge von 15 bis 20 kb. Wenn Sie andere Sequenzierparameter benötigen, lassen Sie es uns gerne wissen! Wir können Ihnen weitere Lösungen anbieten.
Der Output einer PacBio SMRT Cell ist stark abhängig von vielen Faktoren wie der DNA Qualität, dem Vorhandensein von Inhibitoren in der Probe, aber auch der Movie Time. Des Weiteren ist zu beobachten, dass sich Sequenzierlibraries auch bei äquimolarem Pooling gegenseitig hinsichtlich dem generierten Output pro Probe beeinflussen können. Die Gründe dafür sind unzureichend bekannt und auch nicht durch den Technologieprovider PacBio erklärbar. Daher sind die obig genannten Output-Werte als Richtwerte zu verstehen.
Weitere Informationen zu Genome Sequencing
Die Genomsequenzierung wird auch als Whole Genome Sequencing (WGS), Gesamt-Genom-Sequenzierung oder vollständige Genomsequenzierung bezeichnet. Bei dieser Methode wird die gesamte genomische Information sequenziert, um sie in ihrer Gesamtheit zu analysieren. Das bedeutet, dass neben der chromosomalen DNA auch die mitochondriale und, falls vorhanden, die Chloroplasten-DNA sequenziert wird. Whole Genome Sequencing ist somit für eine Vielzahl verschiedener Organismen möglich, darunter Mikroorganismen, Viren, Pflanzen und Säugetiere, wie Menschen oder Mäuse.
Mit der Entwicklung der Hochdurchsatzsequenzierung, die auch als Next-Generation Sequencing bekannt ist, sind die Kosten für Whole Genome Sequencing drastisch gesunken, sodass die Verwendung von Whole Genome Sequencing in der Forschung möglich und erschwinglich wurde. Whole Genome Sequencing hat eine Vielzahl von Anwendungsbereichen und kann z. B. zur Bestimmung der Mutationshäufigkeit im menschlichen Genom eingesetzt werden. Mutationen können auf natürliche Weise entstehen, z. B. durch Replikationsfehler bei der Zellteilung oder durch DNA-Schäden, welche anschließend durch fehleranfällige oder unzureichende Reparaturmechanismen nicht adäquat beseitigt werden. Mutationen können aber auch durch Krankheiten begünstigt werden: Bei Krebserkrankungen ist die Mutationshäufigkeit aufgrund der genomischen Instabilität erhöht. Mit Hilfe von Whole Genome Sequencing können diese veränderten Mutationshäufigkeiten beobachtet werden.
Ein weiterer Anwendungsbereich sind genomweite Assoziationsstudien, bei denen genetische Varianten oder Varianten, die mit einem Phänotyp, assoziiert sind, untersucht werden.
Whole Genome Sequencing ist nicht nur im wissenschaftlichen Bereich eine wichtige Methode. Sie findet auch in der Diagnostik Anwendung und kann zum Beispiel:
- zur Diagnose und Behandlung von Krankheiten, einschließlich seltener Krankheiten,
- zum Nachweis von Krankheitserregern, oder
- zur Nachverfolgung von Krankheitsausbrüchen
eingesetzt werden. Darüber hinaus macht Whole Genome Sequencing die personalisierte Medizin zugänglich und finanzierbar. Sie trägt damit zur Verbesserung des Gesundheitswesens bei. Im Gegensatz zu Whole Exome Sequencing umfasst Whole Genome Sequencing auch die intronischen Regionen eines Genoms und deckt somit alle kodierenden und nicht kodierenden Regionen des Genoms ab. Diese vollständige Sequenzierung des Genoms ermöglicht eine umfassende Analyse des Genoms.
Da Whole Genome Sequencing immer schneller, einfacher und kostengünstiger wird, kann es schon heute einen Beitrag zu Ihrer Forschung leisten!
Unser Engagement für Sie
Schnelle Bearbeitung
Bearbeitungszeit
≤ 15 Werktage
Hohe Qualität
Höchste Genauigkeit bei allen Prozessen
Sichere Datenlieferung
Sichere Bereitstellung der sequenzierten Daten über hausinterne Server
Sichere Aufbewahrung
Sichere Proben- und Datenaufbewahrung nach Projektabschluss
Downloads
Kontaktieren Sie uns
Sie haben noch Fragen oder Interesse an unserem Service? Treten Sie gern mit uns in Kontakt. Wir werden uns schnellstmöglich um Ihr Anliegen kümmern.
Starten Sie Ihr Projekt mit uns
Gerne beraten wir Sie zu unseren Sequenzierdienstleitungen und erarbeiten mit Ihnen gemeinsam die beste Lösung, die auf Ihre klinische Studie oder Forschungsprojekt abgestimmt ist.
Bitte geben Sie, falls möglich, folgende Probeninformationen an: Ausgangsmaterial, Anzahl der Proben, bevorzugte Option für die Vorbereitung der Library, bevorzugte Sequenziertiefe und gewünschte bioinformatische Analysestufe.
