Jede Krebserkrankung ist so individuell wie die Menschen, die davon betroffen sind. Tumoren weisen unterschiedliche genetische Veränderungen auf – selbst bei gleicher Krebsart. Dieses genetische Tumorprofil hat großen Einfluss darauf, welche Therapien wirksam sind – und welche nicht. Deshalb ist eine vollständige molekulargenetische Analyse unerlässlich: Nur so können relevante Veränderungen erkannt und gezielt in die Therapieplanung einbezogen werden. Dadurch profitieren Betroffene von passgenauen Therapieoptionen, während unnötige Behandlungen vermieden werden.
Mit CancerPrecision® erhalten Sie eine vollständige molekulargenetische Tumoranalyse, die alle entscheidenden Informationen erfasst. Die Analyse basiert auf Next-Generation Sequencing (NGS) und ist für alle soliden Tumorentitäten geeignet. CancerPrecision® bildet die umfangreichste Entscheidungsgrundlage für personalisierte Therapien, unabhängig vom Tumorstadium oder der Primärlokalisation.
Untersucht werden über 700 Gene, mehr als 30 therapierelevante Genfusionen sowie zentrale Biomarker wie die Tumormutationslast (TMB), die Mikrosatelliteninstabilität (MSI), den Status der homologen Rekombinationsdefizienz (HRD) und mögliche onkogene virale Infektionen. Ein entscheidender Bestandteil der CancerPrecision®-Analyse ist der Vergleich von Tumor- und Normalgewebe, der die sichere Unterscheidung zwischen somatischen (erworbenen) und keimbahnassoziierten (erblichen) Veränderungen ermöglicht. Nur so lassen sich genetische Befunde korrekt einordnen und Fehleinschätzungen vermeiden. Aufgrund der hohen Sensitivität ermöglicht CancerPrecision® die Detektion von Subklonen und niederfrequenten Resistenzmutationen gegen bestimmte Medikamente. Eine zusätzliche RNA-basierte Fusionsgenanalyse, sowie eine Genexpressionsanalyse können angefordert werden.
Die Ergebnisse werden in einem klar strukturierten medizinischen Befund zusammengeführt. Sie erhalten ein vollständiges molekulargenetisches Tumorprofil samt möglicher Therapieoptionen, damit Sie fundierte und patientenindividuelle Therapieentscheidungen sicher treffen können.
Sie sind in Deutschland versichert? Unsere Kolleginnen und Kollegen vom Zentrum für Humangenetik Tübingen beraten Sie gerne!

CancerPrecision® ist die erste Wahl zur Charakterisierung des Tumors
1 Basierend auf einer qualitativ hochwertigen Probe mit mindestens 20 % Tumorgehalt.
Unser Versprechen an Sie

Service Details
- Tumor- und Normalgewebe-Vergleich für präzise Ergebnisse — mehr erfahren
- Vollständige Sequenzierung und Analyse von mehr als 700 Genen und Fusionen in über 30 Genen — mehr erfahren
- Hohe Sequenzier-Coverage zum Nachweis therapierelevanter subklonaler Varianten: 500-1.000x
- Sensitivität: > 97,6 %*; Spezifität: > 99,9 %
- Analyse der Tumormutationslast (TMB), der Mikrosatelliteninstabilität (MSI) und Virusinfektionen (HPV/EBV/MCV/CMV) — wichtige Biomarker für Immuntherapien — mehr erfahren
- Berechnung des Scores der homologen Rekombinationsdefizienz (HRD) — ein wichtiger Biomarker für PARP-Inhibition — mehr erfahren
- Erkennung von Einzelnukleotidvarianten (SNVs), Insertionen und Deletionen (Indels), Translokationen und Kopienzahlveränderungen (copy number variants, CNVs) — mehr erfahren
- Neben therapierelevanten somatischen (tumorspezifischen) Mutationen werden auch krankheitsverursachende und therapierelevante Keimbahnvarianten berichtet
- Bestimmung von ausgewählten pharmakogenetisch relevanten Keimbahnvarianten, die die Verstoffwechselung bestimmter Krebsmedikamente beeinflussen
- Auflistung aller in Frage kommenden Medikamente mit EMA und/oder FDA-Zulassung, für deren Anwendungsoption entsprechende Biomarker im Tumor nachgewiesen werden konnten — mehr erfahren
- Erfassung von Hinweisen auf CHIP (Klonale Hämatopoese mit unbestimmtem Potenzial)
Optional:
- Multiplex-Immunfluoreszenzfärbung von 9 Markern (CD45, PD-L1, LAG3, p-mTOR, p-ERK, p16, p53, HER2, TROP2) auf derselben Gewebesektion – mehr erfahren
- RNA-basierte Fusionstranskript-Analyse von Tumor-RNA zur Analyse von > 200 Genen (CancerFusionRx®) — mehr erfahren
- Transkriptomsequenzierung von Tumor-RNA, um weitere Erkenntnisse über signifikante Expressionsveränderungen und ihre potenzielle Therapierelevanz zu gewinnen – mehr erfahren
* Basierend auf einer qualitativ hochwertigen Probe mit 20 % Tumorgehalt zum Nachweis einer somatischen heterozygoten Variante.
Beispielbefund
Unsere Standaranforderungen für Proben
Normales Gewebe
- 1–2 ml EDTA-Blut (empfohlene Probenart), oder
- Genomische DNA (1–2 µg)
Tumorgewebe-Optionen
Tumorgehalt mindestens 20 %
- FFPE-Tumorblock (min. Gewebegröße 5x5x5 mm) (empfohlene Probenart)
- Ungefärbte FFPE-Tumorgewebe-Objektträger (min. 10 Schnitte, Gewebegröße 5×5 mm)
- für Färbungen: 5 µm Schnittdicke
- zur Nukleinsäureextraktion (DNA/RNA): 10 µm Schnittdicke
- Isolierte Tumor DNA (> 200 ng)
- frisch gefrorenes Tumorgewebe
- 3x 10 ml cfDNA-Röhrchen für Liquid Biopsy
Hier finden Sie weitere Informationen zum sicheren Versand Ihrer Probe.
Weitere Probenmaterialien
Andere Probenmaterialien sind auf Anfrage möglich. Bitte beachten Sie: Bei unzureichender Probenqualität oder Tumorgehalt kann die Analyse fehlschlagen.
Wenn Sie mehr als eine Option für Tumorproben haben, wenden Sie sich bitte an unseren Diagnostik Support.
Wir unterstützen Sie gerne bei der Auswahl der optimalen Probe für Ihre Patientinnen und Patienten. Für höchste Genauigkeit benötigen wir Tumor- und Normalgewebe für unser somatisches Tumordiagnostik-Panel.
Das macht unseren CancerPrecision®-Service besonders
CancerPrecision® geht weit über eine klassische Paneldiagnostik hinaus: Wir erfassen das vollständige molekulargenetische Tumorprofil – einschließlich aller genetischen Veränderungen, die für das Tumorverhalten, das Therapieansprechen oder mögliche Resistenzmechanismen relevant sind.
Damit Sie sich bestmöglich an den Analyseergebnissen orientieren können, ist der medizinische Bericht klar strukturiert: Die Resultate sind in Hauptkategorien gegliedert, therapieentscheidende Befunde farblich hervorgehoben und alle Inhalte übersichtlich dargestellt. So unterstützen wir Sie dabei, fundierte und individuell passende Therapieentscheidungen zu treffen.
Vergleich von Tumor und Normalgewebe
Das einzig valide Vorgehen zur korrekten Ermittlung somatischer Varianten
Wir sequenzieren stets sowohl die DNA aus dem Tumor als auch aus dem Normalgewebe, in der Regel Blut. Nur so lässt sich sicher unterscheiden, ob eine genetische Veränderung tumorspezifisch oder erblich ist. Diese Unterscheidung ist essenziell, um genetische Veränderungen im Hinblick auf mögliche Therapieansätze korrekt zu bewerten.
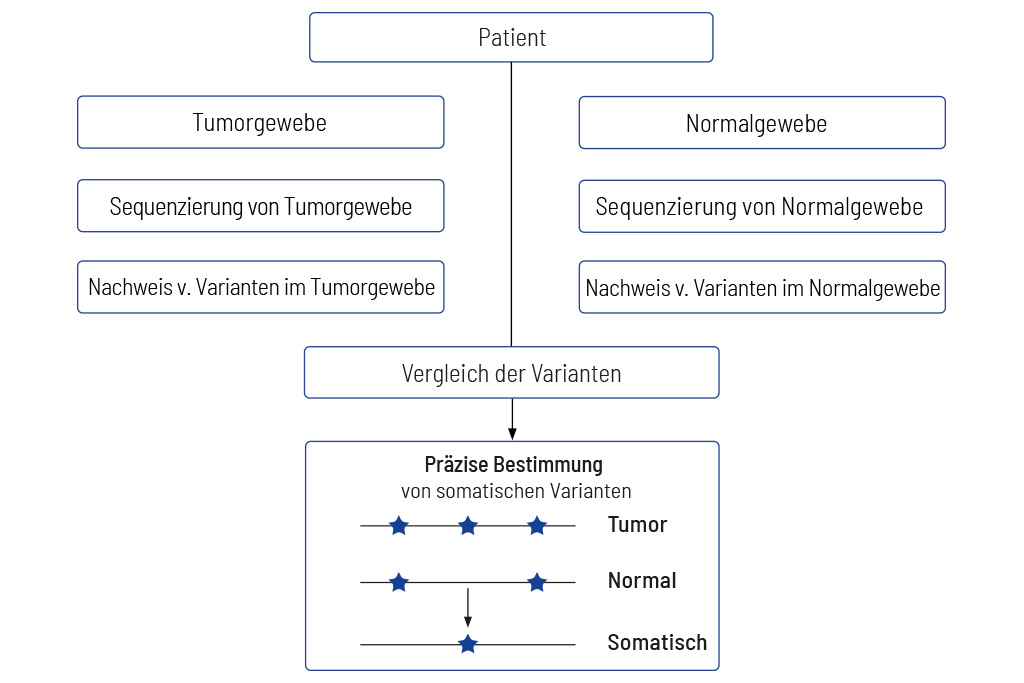
Ohne den Vergleich mit Normalgewebe besteht das Risiko, genetische Veränderungen falsch zuzuordnen – erbliche Mutationen könnten fälschlicherweise als tumorspezifische Veränderungen interpretiert werden oder umgekehrt. Zudem bedeutet das Vorliegen einer vererbten pathogenen Veränderung nicht zwangsläufig, dass sie den Tumor verursacht: Nur der Abgleich von Tumor- und Normalgewebe zeigt, ob eine pathogene erbliche Variante zentraler Treiber des Tumors ist. In der Regel gelingt das durch den Nachweis einer weiteren tumorspezifischen genetischen Veränderung („second hit“) im selben Gen, die das Tumorwachstum fördert.
In bis zu 20% aller Fälle liegt eine erblich bedingte Tumorerkrankung vor – eine wichtige Diagnose für Betroffene und Familienmitglieder, die in einer genetischen Beratung erörtert werden sollte. Der Nachweis von Keimbahnvarianten entscheidet außerdem über die Zulassung bestimmter Medikamente. Deshalb ist es unerlässlich, genetische Veränderungen präzise zuzuordnen.
Eine präzise Bestimmung molekularer Biomarker wie TMB, MSI oder HRD ist nur auf Basis des Vergleichs von Tumor- und Normalgewebe möglich. Ohne diesen Abgleich kann es beispielsweise zu einer Überschätzung der Mutationslast kommen, was zu Wahl ungeeigneter Therapieoptionen führen kann. Patientinnen und Patienten aus in den Populationsdatenbanken unterrepräsentierten Bevölkerungsgruppen profitieren besonders vom Tumor-Normalgewebe Abgleich, da auf unzuverlässige bioinformatische Schätzmodelle zur Unterscheidung von erblichen und tumorspezifischen genetischen Veränderungen verzichtet werden kann.
Genetische Varianten mit therapeutischer Relevanz
Wegweiser für potenziell wirksame Medikamente
Wir analysieren über 700 Gene und identifizieren alle Varianten mit potenzieller therapeutischer Relevanz. Für jede relevante genetische Veränderung stellen wir im Hauptteil des medizinischen Befunds detailliert dar, um welche Art von Veränderung es sich handelt und welche funktionellen Auswirkungen auf die Proteinstruktur oder -funktion zu erwarten sind. Zudem wird die potenzielle therapeutische Relevanz eingeordnet. (A) Diese Informationen bilden die Grundlage für die Diskussion in einem molekularen Tumorboard (MTB).
Ergänzend enthält der Anhang unseres medizinischen Berichts eine therapiebezogene Übersicht (B) für jede identifizierte Veränderung, sofern zugelassene Medikamente (FDA/EMA) vorliegen. Die einzelnen Tabellen fokussieren sich auf konkrete Wirkstoffe, die mit den jeweiligen Varianten in Verbindung stehen. Mit absteigender klinischer Relevanz und ausgewählter Zulassung werden Einschränkungen im Voraus auf ihre Anwendbarkeit geprüft. In dem Beispiel sind zugelassene Medikamente zu sehen, die auf Grund der detektierten Varianten die Zulassungsbeschränkungen für die bestehende Entität erfüllen.
Diese strukturierte Übersicht erleichtert die therapeutische Einordnung der genetischen Befunde erheblich und bietet einen fundierten Ausgangspunkt für die Entscheidung über weiterführende Behandlungsstrategien.
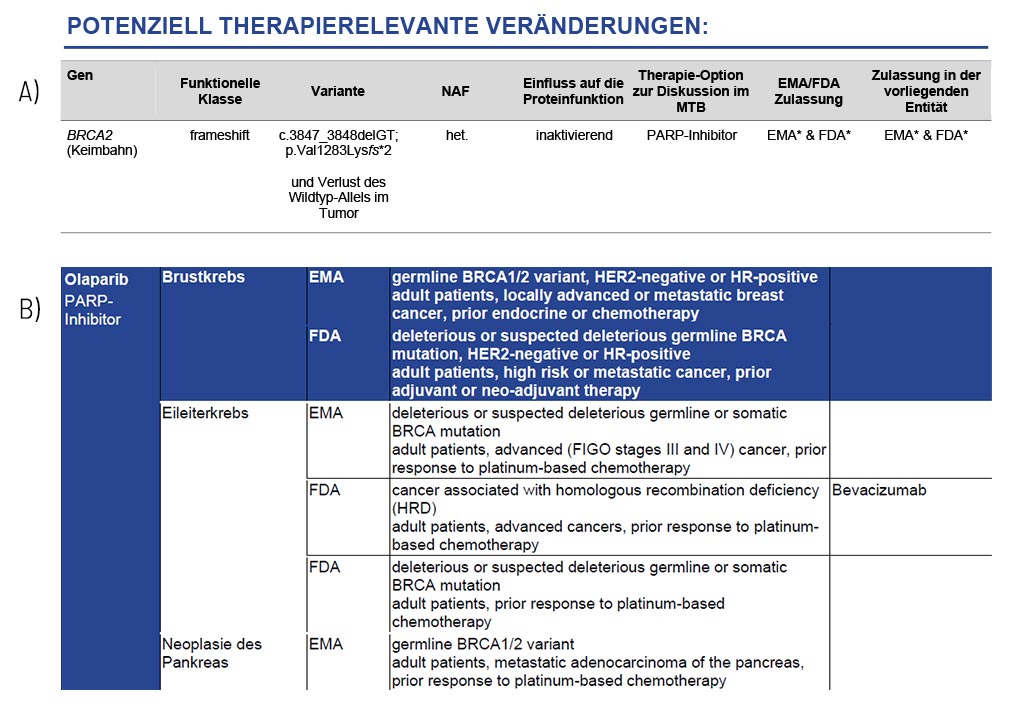
Beispielbefund: Exemplarisch für die befundete HRD– und PIK3CA-Variante und die sich daraus ergebenden therapeutischen Optionen bei einer EMA & FDA Patientin. Oberer Teil (A): Ein Ausschnitt aus Tabelle 1 des Befunds, in dem Varianten mit therapeutischer Relevanz aufgeführt sind. Unterer Teil (B): Ein Auszug der Medikamenten-Auflistung. Neben den abgebildeten Medikamenten werden auch andere Medikamente beschrieben.
Darstellung der Signalwege
Für ein detailliertes Verständnis der veränderten Signalkaskaden
Tumoren entstehen, wenn das Gleichgewicht zwischen Zellwachstum und Zelltod gestört ist. Im Laufe der Tumorentwicklung geraten beide Prozesse zunehmend außer Kontrolle, was zu abnormem Zellwachstum führt.
Unter normalen Bedingungen werden zelluläre Vorgänge durch ein komplexes Netzwerk von Signalwegen genau reguliert. In Tumoren hingegen häufen sich Mutationen in Genen, die zentrale Funktionen in diesen Signalwegen übernehmen. Bereits eine Variante kann mehrere Signalwege beeinflussen – mit direkten Auswirkungen auf das Zellwachstum und mögliche medikamentöse Angriffspunkte.
Neben dem Nachweis krankheitsrelevanter Mutationen ist es daher entscheidend, das Zusammenspiel der betroffenen Signalwege zu verstehen.
Unser medizinischer Bericht gibt Ihnen einen umfassenden Einblick in tumorassoziierte Signalnetzwerke – einschließlich ihrer molekularen Schlüsselakteure, relevanten genetischen Veränderungen und zugehörigen Medikamentenklassen. Er hilft dabei, die komplexen Wechselwirkungen zwischen Signalwegen besser zu verstehen und gezielt auf potenzielle Umgehungsstrategien des Tumors zu reagieren. So lassen sich auch Kombinationstherapien sinnvoll bewerten und in die Therapieplanung einbeziehen.
Berücksichtigte Signalwege
- Signalübertragung über Rezeptor-Tyrosinkinasen
- Zellzyklus
- Reparatur von DNA-Schäden
- Hormonelle Signaltransduktion
- Wnt-Signalweg
- Hedgehog-Signalweg
- Hippo-Signalweg
- Apoptose-Signalweg
- Epigenetische Regulatoren
TMB Bestimmung und MSI Vorhersage
Die Grundlage für therapeutische Entscheidungen über Immuntherapien mit Checkpoint-Inhibitoren
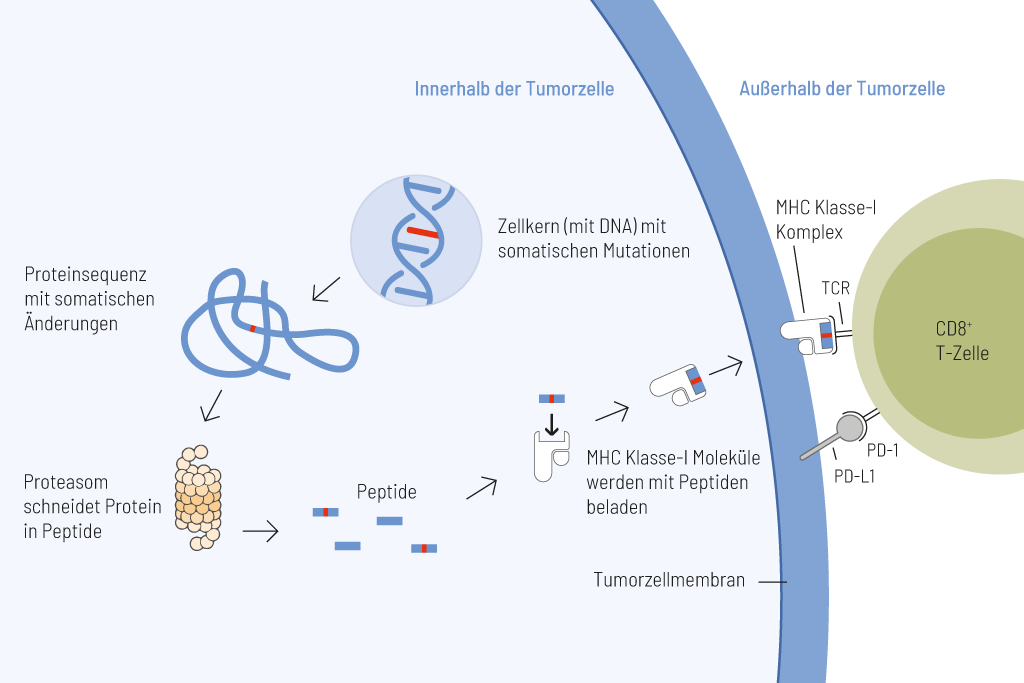
Präsentation von somatischen Peptiden, die von Tumorzellen stammen. Somatische Mutationen treten bei Krebs häufig auf und verändern dauerhaft die genomische Information. Diese genetischen Veränderungen können zur Expression von Proteinen mit veränderten Aminosäuresequenzen führen. Peptide, die eine somatische Veränderung tragen und damit ein besonders starkes immunstimulierendes Potenzial aufweisen, können auf der Oberfläche von Tumorzellen präsentiert werden und eine wirksame Anti-Tumor-Immunantwort hervorrufen.
Die Tumormutationslast (TMB), also die Anzahl der somatischen Mutationen pro Megabase (Mut/Mb), ist ein zulassungsrelevanter Biomarker und prädiktiv für das Ansprechen auf eine Behandlung mit Immun-Checkpoint-Inhibitoren.
Je höher die Anzahl der genetischen Veränderungen innerhalb einer Tumorzelle ist, desto mehr mutierte Proteine werden gebildet. Diese mutierten Proteine werden zu kurzen Fragmenten (Oligopeptiden) verarbeitet, die auf der Zelloberfläche von Tumorzellen präsentiert werden. Solche mutierten Peptide werden als Neoantigene bezeichnet und sind hoch immunogen. Das bedeutet, dass sie von Immunzellen, insbesondere von T-Zellen, sehr effektiv erkannt werden. Bestimmte T-Zellen sind in der Lage, Tumorzellen nach der Antigenerkennung direkt zu eliminieren. Je höher die Zahl der Mutationen ist, desto größer ist daher die Chance, dass Neoantigene auf Tumorzellen präsentiert werden, und desto effizienter ist die Tumorbekämpfung durch T-Zellen. Die Aktivität von T-Zellen kann jedoch durch Immun-Checkpoints unterdrückt werden. Daher sind Therapien, die auf diese Checkpoints abzielen (sogenannte Immun-Checkpoint-Inhibitoren), besonders wirksam bei Tumoren mit hoher Mutationslast.4
Durch den Vergleich von Tumor- und Normalgewebe können wir den TMB präzise berechnen. In unserem medizinischen Befund liefern wir nicht nur die TMB-Klassifikation, sondern auch den exakten Mutationswert pro Megabase. Dadurch bieten wir eine deutlich differenziertere Einschätzung als die einfache Einteilung in ‚hoch‘ oder ‚niedrig‘. Besonders in Grenzfällen ermöglicht diese präzise Bestimmung eine fundiertere Beurteilung, ob eine Immuntherapie infrage kommt.
Die Genauigkeit der TMB-Berechnung hängt wesentlich von der Größe des analysierten Genpanels ab. Mit 2,96 Mb liegt CancerPrecision® deutlich über der empfohlenen Mindestgröße von 1,5 Mb und gewährleistet eine robuste Berechnung der Mutationslast.
Zusätzlich bestimmen wir den MSI-Status (Mikrosatelliteninstabilität), einen weiteren wichtigen Marker für das Immuntherapie-Ansprechen. Mikrosatelliten sind kleine repetitive DNA-Sequenzen, die sich im gesamten Genom finden. Die Größe von Mikrosatelliten kann sich aufgrund von Ausfällen der DNA-Mismatch-Reparaturmechanismen verändern, wenn die zugrundeliegenden Gene durch Mutationen beeinträchtigt sind. Hierdurch entstehen gehäuft frameshift Mutationen, die die Mutationslast stark erhöhen.
Neben klassischer PCR-Methoden erfolgt die Vorhersage bei uns ebenfalls vollständig NGS-basiert. Dadurch können deutlich mehr Mikrosatellitenregionen analysiert werden – ohne, daß es einer gesonderten Laboranalyse bedarf, wird die Berechnung für jede sequenzierte Tumor-Probe durchgeführt. Diese Berechnung wurde mit Hunderten von Probenpaaren aus Normal- und Tumorgewebe verschiedener Tumorarten, bei denen mehr als 2.500 Mikrosatelliten-Loci untersucht wurden, validiert. Zusätzlich bieten wir auf Wunsch natürlich auch das klassische, PCR-basierte Verfahren für die MSI Bestimmung an.
HRD – Homolge Rekombinationsdefizienz
Verschiedene DNA-Reperaturmechanismen sorgen in gesunden Zellen für ein stabiles und fehlerfreies Genom. Die homologe Rekombination (HR) ist hierbei für die Reparatur von DNA-Schäden, die beide DNA-Stränge betreffen (Doppelstrangbrüche), von besonderer Bedeutung. Ist dieser Mechanismus gestört sammeln sich Mutationen, Chromosomenaberrationen und andere Fehler im Genom an und man spricht von einer homologen Rekombinationsdefizienz (HRD). Durch die daraus resultierende genomische Instabilität begünstigt die HRD die Tumorentwicklung und trägt zur Tumorentstehung bei verschiedenen Tumorarten, insbesondere bei Brust- und Eierstockkrebs, bei.5, 6
Für zahlreiche HR-defiziente Tumoren gibt es wirksame therapeutische Ansätze, die sich die sogenannte synthetische Letalität zunutze machen, beispielsweise durch Verabreichung von PARP-Inhibitoren und platinhaltigen Chemotherapien. Diese Therapeutika verursachen DNA-Brüche, welche die verbleibende DNA-Reparaturmaschinerie von HR-defizienten Tumoren so stark belasten, dass diese absterben, während gesunde Zellen dank intakter Reperaturmechanismen überleben. Zur Identifizierung der Tumoren, bei denen diese Medikamente eingesetzt werden können, ist eine zuverlässige Bestimmung des HRD-Status von höchster Relevanz.
HR-defiziente Tumoren werden häufig durch Keimbahn oder somatische Mutationen in BRCA1 oder BRCA2 verursacht. Daher wurde dieses Muster früher als BRCAness bezeichnet. Darüber hinaus haben sich Mutationen in anderen HR-Genen wie RAD51C, ATM und PALB2 als mögliche Ursache für eine HRD erwiesen. Allerdings muss nicht jeder genetische Defekt in HR-Genen zwangsläufig zu einer HRD im Tumor führen. Zudem kann eine HRD auch dann vorliegen, wenn keine HR-Genmutation durch Sequenzierung nachweisbar ist (z. B. wurde eine Promotor-Methylierung von BRCAness-Genen ebenfalls als Ursache für eine HRD identifiziert). Werden ausschließlich Sequenz-Mutationen in BRCAness-Genen untersucht, kann eine potenzielle HRD unentdeckt bleiben. Um sicherzustellen, dass HR-defiziente Tumoren nicht übersehen werden, berechnen wir den HRD-Score als Teil jeder CancerPrecision®- Analyse – unabhängig von der Tumorentität.
Der HRD-Score misst eine spezielle Signatur der genomischen Instabilität anhand der Anzahl der Chromosomenveränderungen wie Insertionen, Deletionen und Substitutionen, die auf genomweiter Ebene auftreten, auch ohne, dass die dafür verantwortlichen Genmutationen identifiziert werden müssen. Diese Signatur wird anschließend zur Berechnung des HRD-Scores der Tumorprobe verwendet. Der HRD-Score wird aus drei typischen HRD-Ereignissen berechnet:
- Loss of heterozygosity (LOH)
- Telomeric allelic imbalance (TAI)
- Large-scale state transition (LST)
Der HRD-Score wird in unserem CancerPrecision®- Befund zusammen mit allen identifizierten somatischen Mutationen und ausgewählten Genfusionen sowie TMB, MSI und CNVs angegeben.
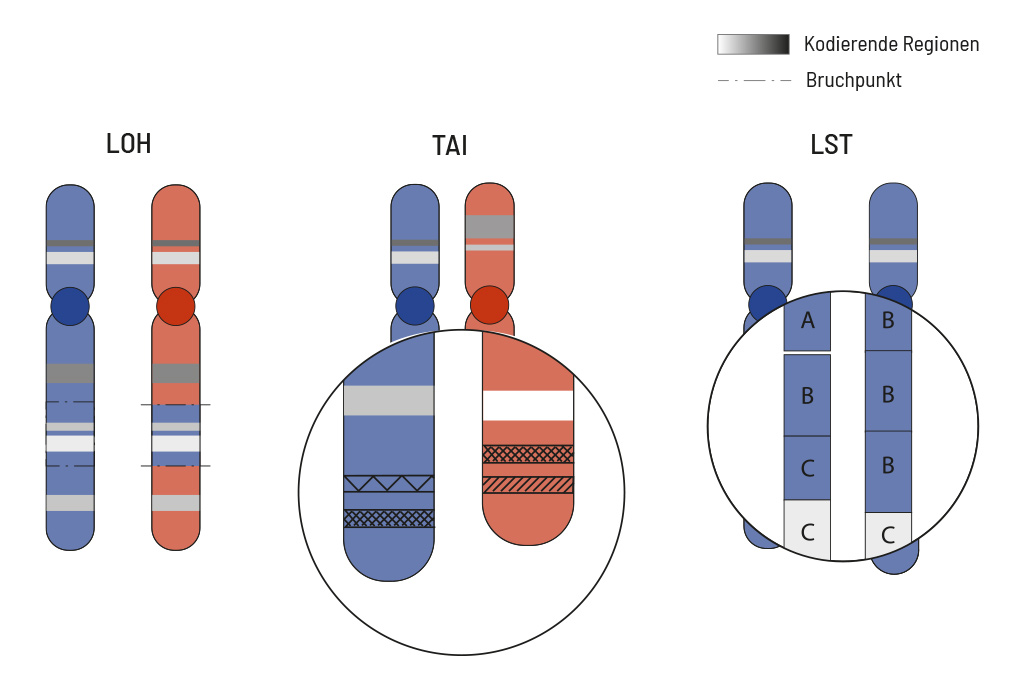
LOH ist der irreversible Verlust eines elterlichen Allels, welcher besonders schwerwiegend ist, wenn das andere Allel bereits inaktiviert ist. LOH-Regionen werden definiert als Regionen, die größer als 15 Mb, aber kleiner als das gesamte Chromosom sind. Eine TAI tritt auf, wenn das Telomer eines Chromosoms in einem der beiden Chromosomen stark verkürzt ist, wodurch ein allelisches Ungleichgewicht in dieser Region entsteht. Dieses Ungleichgewicht resultiert daraus, dass die repetitiven DNA-Sequenzen in Telomerregionen besonders anfällig für HRD sind. Zur Bestimmung der LST wird die Anzahl der Bruchpunkte zwischen benachbarten Chromosomenregionen bewertet, die Kopienzahlgewinne oder -verluste von mehr als 10 Mb umfassen.
CNV-Analyse
Bestimmung von Deletionen/Amplifikationen für die höchste therapeutische Ausbeute
CNVs (Copy Number Variants) spielen in der Tumorgenetik oft eine wichtige Rolle. Die Kenntnis der Veränderungen bei CNVs hilft bei der Wahl der optimalen Behandlung. Die CNV-Analyse ist daher bei uns ein integraler Bestandteil der somatischen Tumordiagnostik.
Zelluläre Prozesse sind streng reguliert. Diese Regulierung hängt von der korrekten Funktion der Gene ab. Bei Tumoren ist die Kopienzahl der Gene häufig verändert, wodurch die korrekte Funktion der betroffenen Gene beeinträchtigt wird. Eine Erhöhung der Kopienzahl eines Gens kann dessen Aktivität steigern, während eine (teilweise) Deletion zu einem Funktionsverlust führen kann. Daher können Chromosomenaberrationen, die zu Veränderungen der Kopienzahl führen, auch therapeutische Konsequenzen haben.
Bei Tumoren sind Kopienzahlveränderungen (CNVs) aufgrund der allgemeinen genomischen Instabilität häufig. Hier sind oft große Chromosomenteile entweder deletiert oder amplifiziert. Es ist wichtig, diese Deletionen/Amplifikationen zu verstehen und die Gene in der betroffenen Region mit therapeutischer Relevanz zu kennen. Daher ermitteln wir Deletionen und Amplifikationen anhand der NGS-Daten.
Zusammen mit den betroffenen Genen mit therapeutischer Relevanz werden die Deletionen und Amplifikationen zu Beginn des Befunds aufgelistet. Ein vollständiges CNV-Profil der analysierten Regionen ist im Anhang des Befunds zu finden.
CancerIFP
Multiplex-Immunfluoreszenzfärbung zur Analyse von Immun- und Onkoproteinen
Die klassische Immunhistochemie (IHC) erlaubt den Nachweis eines einzelnen Markers pro Gewebeschnitt. Häufig steht nur wenig Gewebe zur Verfügung, und es können nur einzelne Marker untersucht werden. CancerIFP hingegen ermöglicht die gleichzeitige Analyse mehrerer Marker auf einem einzigen Tumorgewebeschnitt.
Für jeden Marker wird ein spezifischer, fluoreszenzmarkierter Antikörper eingesetzt, der gezielt an sein Zielprotein bindet. Beim Auslesen des gefärbten Schnitts werden die Fluoreszenzsignale digital erfasst und getrennt dargestellt. So entsteht ein präzises Bild der Proteinexpression und ihrer räumlichen Verteilung im Tumorgewebe.
CancerIFP liefert Einblicke in relevante Immun-Checkpoints und tumorspezifische Signalwege. Die Analyse umfasst neun Marker, die gemeinsam auf einem FFPE-Gewebeschnitt untersucht werden. Alle Analysen werden auf unserer Whole-Slide-Immunfluoreszenzplattform durchgeführt und durch eine Fachärztin für Neuropathologie oder einen Facharzt für Pathologie bewertet. Die Analyse wurde bei uns im Haus als Laboratory-Developed Test (LDT) entwickelt. Jeder Antikörper wurde hausintern validiert, und die Bewertung folgt einem konsistenten Scoringsystem, das sich an etablierten IHC-Standards orientiert. Die Ergebnisse liefern ein detailliertes Bild der Proteinexpression im Tumorgewebe und ergänzen unsere umfangreichen Tumordiagnostik-Ergebnisse um Befunde auf Proteinebene.
CancerFusionRx®
RNA-basierte Identifizierung von Fusionstranskripten
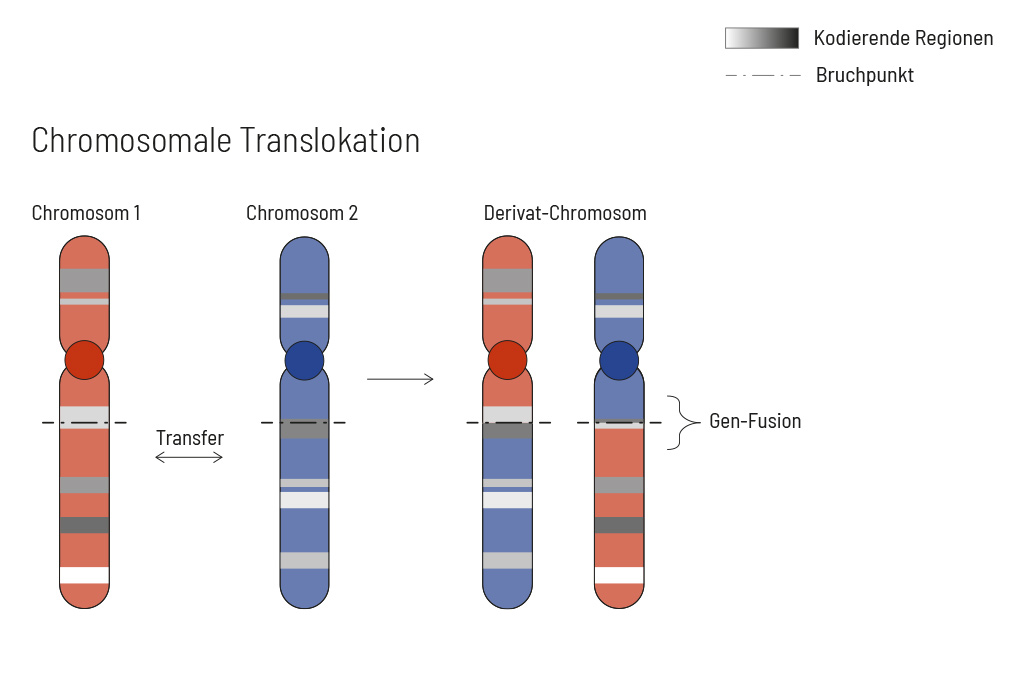
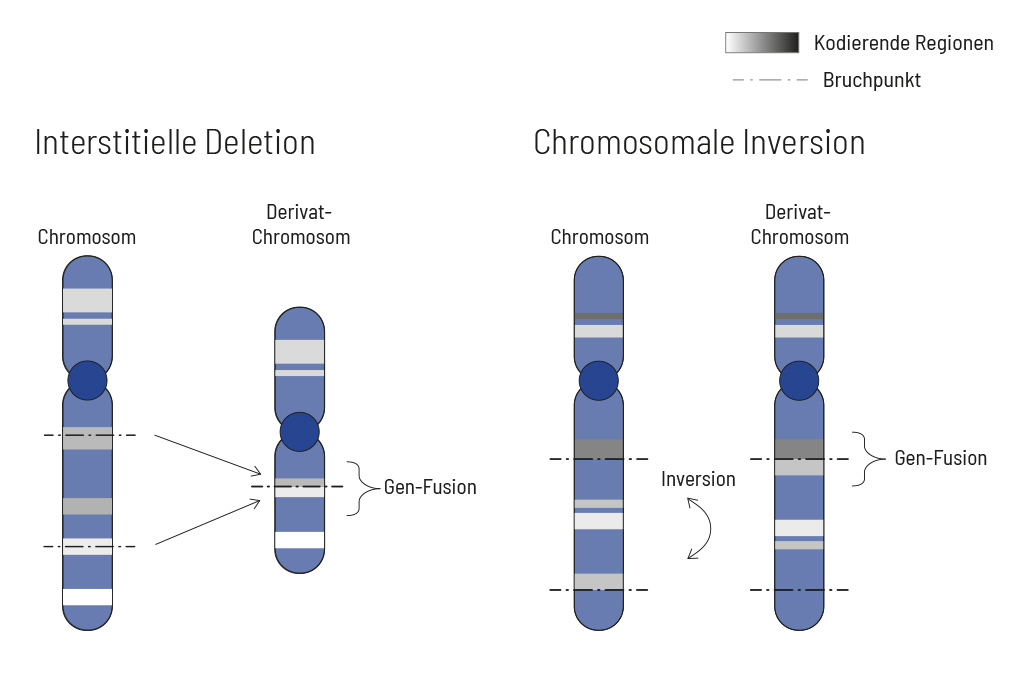
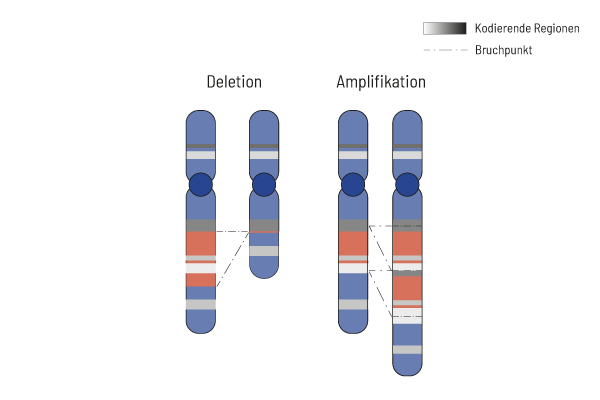
Chromosomale Veränderungen treten häufig und unabhängig von der Tumorentität auf. Infolgedessen kann es zu Genfusionen im Tumorgenom kommen. Fusionen sind oftmals starke Treiber der Tumorerkrankung und daher für Behandlungsentscheidungen sehr wichtig. Herkömmliche, auf PCR-Technologie basierende Methoden, können eine Fusion nicht nachweisen, wenn der Fusionspartner nicht bekannt ist (dies ist bei NTRK-Fusionen häufig der Fall). Selbst Analysen des gesamten Transkriptoms sind nicht sensitiv genug, insbesondere wenn der Tumorgehalt niedrig ist. Um alle bekannten und zuvor beschriebenen, sowie neuartigen Genfusionen mit einer therapeutischen Option nachzuweisen, haben wir eine gezielte Anreicherung auf RNA-Basis entwickelt.
Das Design umfasst aktuell mehr als 200 Gene für den Nachweis von Fusionen und beinhaltet über 132 Exon-Exon spezifische Anreicherungen mit bekannten Bruchpunkten. Diese Methode ist den DNA-basierten Methoden und auch Ansätzen auf Transkriptom-Basis überlegen. Wir empfehlen dringend, die genetische Tumordiagnostik durch RNA-Anreicherung für Fusionen zu ergänzen, um ein möglichst vollständiges Verständnis der Tumorbiologie zu erhalten.
Genexpressionsanalyse
Tumor RNA Analyse für weitere Erkenntnisse
Die Genexpressionsanalyse ist entscheidend für ein tiefergehendes Verständnis von festen Tumoren auf molekularer Ebene. Sie liefert zusätzliche Informationen für Diagnose, Prognose und personalisierte Behandlung.
Mit dieser Zusatzleistung führen wir zusätzliche Transkriptom-Sequenzierungen durch, um die Genexpression aller tumorrelevanten Gene zu ermitteln. Die abweichende Genexpression wird durch den Vergleich mit den Expressionsmustern aus unserer umfangreichen hauseigenen Kohorte von Tumortranskriptomen bestimmt. Die Ergebnisse können DNA-Befunde auf RNA-Ebene verifizieren, Empfehlungen für weitere Analysen (z. B. IHC-Färbung) geben und potenzielle therapeutische Ziele identifizieren, selbst wenn keine DNA-Mutationen nachweisbar sind.
Die Ergebnisse werden in den CancerPrecision®-Bericht einbezogen und sowohl in Tabellenform (mit den relevanten Expressionswerten, z. B. FPKM pro Inhouse-Kohorte, Tabelle A) als auch in Form von Violinplots dargestellt, um weiteren visuellen Kontext zu schaffen (Abbildung B). Mögliche therapeutische Strategien werden gegebenenfalls auch in der umfangreichen Medikamentenliste im Abschnitt „Ergänzungen“ aufgeführt.
A)

B)


Die X-Achse zeigt die Genexpression in FPKM in logarithmischer Skala. Der rote Balken markiert das Niveau der Genexpression des untersuchten Gens in der analysierten Probe. Die Unterteilung der Violinplots durch Längsbalken stellt die Perzentilverteilung dar (Perzentile von links nach rechts: 0-25, 25-50, 50-75, und 75-100). Jede Abbildung gibt die Verteilung von oben nach unten wieder: 1) Die Verteilung der Genexpression des angegebenen Gens in einer Kohorte von Tumorproben, die der Tumorentität des Patienten entsprechen. 2) Die Verteilung der Genexpression des angegebenen Gens in einer Kohorte von Tumorproben mit anderen Tumorentitäten. 3) Die Verteilung der Genexpression aller exprimierten Gene in der analysierten Tumor-RNA des Patienten.
Genverzeichnis
Genliste für DNA-basierte Analyse (787 Gene, CancerPrecision®, TUM01)
ABCB1, ABCG2, ABL1, ABL2, ABRAXAS1, ACD, ACVR1, ACVR2A, ADGRA2, ADRB1, ADRB2, AIP, AIRE, AJUBA, AKT1, AKT2, AKT3, ALK, ALOX12B, AMER1, ANKRD26, APC, APLNR, APOBEC3A, APOBEC3B, AR, ARAF, ARFRP1, ARHGAP35, ARID1A, ARID1B, ARID2, ARID5B, ASXL1, ASXL2, ATM, ATR, ATRX, AURKA, AURKB, AURKC, AXIN1, AXIN2, AXL, B2M, B4GALNT1, BAP1, BARD1, BAX, BCHE, BCL10, BCL11A, BCL11B, BCL2, BCL2L1, BCL2L11, BCL3, BCL6, BCL9, BCOR, BCORL1, BCR, BIRC2, BIRC3, BIRC5, BLM, BMI1, BMPR1A, BRAF, BRCA1, BRCA2, BRD3, BRD4, BRD7, BRIP1, BTK, BTN3A1, BUB1B, CACNA1S, CALR, CARD11, CASP8, CBFB, CBL, CBLB, CBLC, CCDC6, CCND1, CCND2, CCND3, CCNE1, CD274, CD276, CD70, CD79A, CD79B, CD82, CDC42, CDC73, CDH1, CDH11, CDH2, CDH3, CDH5, CDK1, CDK12, CDK2, CDK4, CDK5, CDK6, CDK8, CDKN1A, CDKN1B, CDKN1C, CDKN2A, CDKN2B, CDKN2C, CEACAM5, CEBPA, CENPA, CEP57, CFTR, CHD1, CHD2, CHD4, CHEK1, CHEK2, CIC, CIITA, CLDN18, CNKSR1, COL1A1, COMT, COQ2, CREB1, CREBBP, CRKL, CRLF2, CRTC1, CSF1R, CSF3R, CSMD1, CSNK1A1, CTAG1B, CTCF, CTLA4, CTNNA1, CTNNB1, CTR9, CTRC, CUL3, CUX1, CXCR4, CYLD, CYP1A2, CYP2A7, CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5, CYP4F2, DAXX, DCC, DDB2, DDR1, DDR2, DDX11, DDX3X, DDX41, DHFR, DICER1, DIS3L2, DLL3, DNMT1, DNMT3A, DOT1L, DPYD, E2F3, EED, EFL1, EGFR, EGLN1, EGLN2, EIF1AX, ELAC2, ELF3, EME1, EML4, EMSY, EP300, EPAS1, EPCAM, EPHA2, EPHA3, EPHB4, EPHB6, ERBB2, ERBB3, ERBB4, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERG, ERRFI1, ESR1, ESR2, ETNK1, ETV1, ETV4, ETV5, ETV6, EWSR1, EXO1, EXT1, EXT2, EZH1, EZH2, EZHIP, F3, FAN1, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FAS, FAT1, FBXO11, FBXW7, FEN1, FES, FGF10, FGF14, FGF19, FGF2, FGF23, FGF3, FGF4, FGF5, FGF6, FGF9, FGFR1, FGFR2, FGFR3, FGFR4, FH, FLCN, FLI1, FLT1, FLT3, FLT4, FOLH1, FOLR1, FOXA1, FOXE1, FOXL2, FOXO1, FOXQ1, FRK, FRS2, FUS, FYN, G6PD, GALNT12, GATA1, GATA2, GATA3, GATA4, GATA6, GGT1, GLI1, GLI2, GLI3, GNA11, GNA13, GNAQ, GNAS, GNB3, GPC3, GPER1, GREM1, GRIN2A, GRM3, GSK3A, GSK3B, GSTP1, H3-3A, H3-3B, H3C1, H3C2, H3C3, HABP2, HAVCR2, HCK, HDAC1, HDAC2, HDAC6, HGF, HIF1A, HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, HLA-DRB1, HMGA2, HMGCR, HMGN1, HNF1A, HNF1B, HOXB13, HRAS, HSD3B1, HSP90AA1, HSP90AB1, HTR2A, ICOSLG, ID2, ID3, IDH1, IDH2, IDO1, IFNGR1, IFNGR2, IFNL3, IGF1, IGF1R, IGF2, IGF2R, IKBKB, IKBKE, IKZF1, IKZF3, IL1B, IL1RN, IL7R, INPP4A, INPP4B, INPPL1, INSR, IRF1, IRF2, IRS1, IRS2, IRS4, ITPA, JAK1, JAK2, JAK3, JUN, KAT6A, KDM5A, KDM5C, KDM6A, KDR, KEAP1, KIAA1549, KIF1B, KIT, KLF2, KLF4, KLHL6, KLLN, KMT2A, KMT2B, KMT2C, KMT2D, KRAS, KSR1, LAG3, LAMP1, LATS1, LATS2, LCK, LIG4, LIMK2, LRP1B, LRRK2, LTK, LYN, LZTR1, MAD2L2, MAF, MAGEA1, MAGEA12, MAGEA3, MAGEA4, MAGEA8, MAGI1, MAGI2, MAML1, MAP2K1, MAP2K2, MAP2K3, MAP2K4, MAP2K5, MAP2K6, MAP2K7, MAP3K1, MAP3K13, MAP3K14, MAP3K3, MAP3K4, MAP3K6, MAP3K8, MAPK1, MAPK11, MAPK12, MAPK14, MAPK3, MAX, MBD4, MC1R, MCL1, MDC1, MDH2, MDM2, MDM4, MECOM, MED12, MEF2B, MEN1, MERTK, MET, MGA, MGMT, MITF, MLH1, MLH3, MLLT10, MLLT3, MMP2, MMS22L, MN1, MPL, MRE11, MS4A1, MSH2, MSH3, MSH4, MSH5, MSH6, MSLN, MSR1, MST1R, MTAP, MTHFR, MTOR, MT-RNR1, MTRR, MUC1, MUTYH, MXI1, MYB, MYC, MYCL, MYCN, MYD88, MYH11, MYH9, MYOD1, NAT2, NBN, NCOA1, NCOA3, NCOR1, NF1, NF2, NFE2L2, NFKB1, NFKB2, NFKBIA, NFKBIE, NIN, NKX2-1, NLRC5, NOTCH1, NOTCH2, NOTCH3, NOTCH4, NPM1, NQO1, NR1I3, NRAS, NRG1, NSD1, NSD2, NSD3, NT5C2, NTHL1, NTRK1, NTRK2, NTRK3, NUDT15, NUMA1, NUP98, NUTM1, OBSCN, OPRM1, PAK1, PAK3, PAK4, PAK5, PALB2, PALLD, PARP1, PARP2, PARP4, PAX3, PAX5, PAX7, PBK, PBRM1, PBX1, PDCD1, PDCD1LG2, PDGFA, PDGFB, PDGFC, PDGFD, PDGFRA, PDGFRB, PDK1, PDPK1, PGR, PHF6, PHOX2B, PIAS4, PIGA, PIK3C2A, PIK3C2B, PIK3C2G, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PIK3R1, PIK3R2, PIK3R3, PIM1, PLCG1, PLCG2, PLK1, PMEL, PML, PMS1, PMS2, POLB, POLD1, POLE, POLH, POLQ, POR, POT1, PPARG, PPM1D, PPP2R1A, PPP2R2A, PRAME, PREX2, PRKAR1A, PRKCA, PRKCI, PRKDC, PRKN, PRMT5, PRR4, PSMB1, PSMB10, PSMB2, PSMB5, PSMB8, PSMB9, PSMC3IP, PSME1, PSME2, PSME3, PTCH1, PTCH2, PTEN, PTGS2, PTK2, PTK7, PTPN11, PTPN12, PTPRC, PTPRD, PTPRS, PTPRT, RABL3, RAC1, RAC2, RAD21, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD54B, RAD54L, RAF1, RALGDS, RARA, RASA1, RASAL1, RB1, RBM10, RECQL4, REST, RET, RFWD3, RFX5, RFXANK, RFXAP, RHBDF2, RHEB, RHOA, RICTOR, RIF1, RINT1, RIPK1, RIT1, RNASEL, RNF43, ROS1, RPS20, RPS6KB1, RPS6KB2, RPTOR, RSF1, RSPO1, RSPO2, RSPO3, RSPO4, RUNX1, RYR1, SAMHD1, SAV1, SBDS, SCG5, SDHA, SDHAF2, SDHB, SDHC, SDHD, SEC23B, SERPINB9, SETBP1, SETD2, SETDB1, SF3B1, SGK1, SH2B3, SHH, SHLD2, SIK2, SKP2, SLC19A1, SLC26A3, SLC45A2, SLCO1B1, SLFN11, SLIT2, SLX4, SMAD3, SMAD4, SMARCA2, SMARCA4, SMARCB1, SMARCE1, SMC1A, SMC3, SMO, SOCS1, SOS1, SOX11, SOX2, SOX9, SPEN, SPINK1, SPOP, SPRED1, SRC, SRD5A2, SRGAP1, SRSF2, SSTR2, SSX1, STAG2, STAT1, STAT3, STAT5A, STAT5B, STK11, SUCLG2, SUFU, SUZ12, SYK, TACSTD2, TAF1, TAF15, TAP1, TAP2, TAPBP, TBK1, TBX3, TCF3, TCF4, TCL1A, TEK, TERC, TERF2IP, TERT, TET1, TET2, TFE3, TGFB1, TGFBR2, TMEM127, TMPRSS2, TNFAIP3, TNFRSF13B, TNFRSF14, TNFRSF8, TNFSF11, TOP1, TOP2A, TP53, TP53BP1, TP63, TPMT, TPX2, TRAF2, TRAF3, TRAF5, TRAF7, TRIM28, TRRAP, TSC1, TSC2, TSHR, TTK, TYMS, U2AF1, UBE2T, UBR5, UGT1A1, UGT2B15, UGT2B7, UIMC1, USP9X, VEGFA, VEGFB, VHL, VKORC1, VTCN1, WRN, WT1, XIAP, XPA, XPC, XPO1, XRCC1, XRCC2, XRCC3, XRCC5, XRCC6, YAP1, YES1, ZFHX3, ZNF217, ZNF703, ZNRF3, ZRSR2
DNA-basierte Detektion von ausgewählten strukturellen Veränderungen in den Genen
ALK, BCL2, BCOR, BCR, BRAF, BRD4, CDKN2A, CDKN2B, EGFR, ERG, ETV4, ETV6, EWSR1, FGFR1, FGFR2, FGFR3, FUS, MET, MSH2, MYB, MYC, NFE2L2, NOTCH2, NRG1, NTRK1, NTRK2, NTRK3, PAX3, PDGFB, RAF1, RARA, RET, ROS1, SSX1, SUZ12, TAF15, TCF3, TFE3, TMPRSS2
Genliste für RNA-basierte Fusionstranskriptanalyse (CancerFusionRx®, STR01)
Genliste für de-novo Fusionstranskripterkennung
ABL1, ACTB, AFAP1, AGK, AKAP4, AKAP9, AKAP12, AKT1, AKT2, AKT3, ALK, ARHGAP6, ARHGAP26, ASPL, ASPSCR1, ATF1, ATP1B1, ATRX, AVIL, AXL, BAG4, BCL2, BCOR, BCORL1, BCR, BEND2, BICC1, BRAF, BRD3, BRD4, c11orf95, CAMTA1, CCAR2, CCDC6, CCDC88A, CCDC170, CCNB3, CCND1, CD44, CD74, CEP85L, CIC, CLDN18, CLIP1, CLTC, CNTRL, COL1A1, CREB1, CREB3L1, CREB3L2, CRTC1, CTNNB1, DDIT3, DNAJB1, EGFR, EML4, EPC1, EPCAM, ERBB2, ERBB4, ERG, ESR1, ESRRA, ETV1, ETV4, ETV5, ETV6, EWSR1, EZR, FEV, FGFR1, FGFR2, FGFR3, FLI1, FN1, FOXO1, FOXO4, FOXR2, FUS, GLI1, GOPC, GPR128, HEY1, HMGA2, HTRA1, IGF1R, INSR, JAK2, JAZF1, KIAA1549, KIF5B, KIT, LEUTX, LMNA, LPP, LTK, MAGI3, MAML1, MAML2, MAML3, MAMLD1, MAP3K8, MARS1, MAST1, MAST2, MEAF6, MET, MGA, MGMT, MITF, MKL2, MN1, MSH2, MYB, MYBL1, MYC, NAB2, NCOA1, NCOA2, NCOA3, NCOA4, NFATC2, NFIB, NOTCH2, NPM1, NR4A3, NRG1, NRG2, NSD3, NTRK1, NTRK2, NTRK3, NUTM1, PAX3, PAX7, PAX8, PBX1, PDGFB, PDGFD, PDGFRA, PDGFRB, PHF1, PIK3CA, PLAG1, PML, POU5F1, PPARG, PPARGC1A, PPP1CB, PRKACA, PRKAR1A, PRKCA, PRKCB, PRKD1, PRKD2, PRKD3, PTPRZ1, QKI, RAD51B, RAF1, RANBP2, RARA, RELA, RELCH, RET, ROS1, RPS6KB1, RREB1, RSPO2, RSPO3, SDC1, SDC4, SH3PXD2A,SLC1A2, SHTN1, SLC34A2, SLC44A1, SLC45A3, SND1, SQSTM1, SS18, SSX1, SSX2, SSX4, STAT6, STRN, SUZ12, TACC1, TACC2, TACC3, TAF2N, TAF15, TCF3, TCF12, TERT, TFE3, TFEB, TFG, THADA, TMPRSS2, TPM3, TPR, TRIM24, TRIM33, TRIO, TTYH1, VGLL2, VGLL3, VMP1, WT1, WWTR1, YAP1, YWHAE, ZC3H7B, ZMYM2, ZNF703
Genliste für ausgewählte Bruchpunkte in diesen Fusiongenen
AFAP1-NTRK2, ATP1B1-NRG1, BCOR-CCNB3, BRD3-NUTM1, BRD4-NUTM1, CCDC6–RET, CCDC88A-ALK, CD74-NRG1, CD74-ROS1, CLTC-ALK, DNAJB1-PRKACA, EGFR-PPARGC1A, EML4-ALK, ETV6 NTRK2, ETV6-NTRK3, EWSR1-ATF1, EWSR1-ERG, EWSR1-FLI1, EWSR1-WT1, EZR-ROS1, FGFR1-TACC1, FGFR2-BICC1, FGFR2 TACC3, FGFR3-TACC3, KIAA1549-BRAF, KIF5B-ALK, KIF5B–RET, MGA-NUTM1, NAB2-STAT6, NCOA4–RET, NPM1-ALK, NSD3-NUTM1, PAX3-FOXO1, PAX7-FOXO1, PPP1CB-ALK, PRKAR1A-RET, QKI-NTRK2, RANBP2- ALK, RPS6KB1–VMP1, SDC4-NRG1, SDC4 ROS1, SLC34A2-ROS1, SND1-BRAF, SS18-SSX1, SS18-SSX2, STRN ALK, TMPRSS2-ERG, TPM3-ALK, TPM3-NTRK1, TPM3-ROS1, TPR NTRK1, TRIM24-BRAF, TRIM24-NTRK2, TRIM33-RET, TRIO-TERT
Liste für spezifische Transkriptvarianten
EGFR del ex2-22 (mLEEK), EGFR del ex25-26 (EGFRvIVb), EGFR del ex25-27 (EGFRvIVa), EGFR del ex26-27, EGFR del ex14-15 (vII), EGFR del ex2-7 (vIII), FGFR2IIIb, MET ex14 skipping, NFE2L2 ex2 skipping, PDGFRA del ex8-9
Referenzen
1 Jones, S. et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Science translational medicine 7, 283ra53; 10.1126/scitranslmed.aaa7161. (2015).
2 Sun, J. X. et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS computational biology 14, e1005965; 10.1371/journal.pcbi.1005965 (2018).
3 Nassar, A. H. et al. Ancestry-driven recalibration of tumor mutational burden and disparate clinical outcomes in response to immune checkpoint inhibitors. Cancer cell 40, 1161-1172.e5; 10.1016/j.ccell.2022.08.022 (2022).
4 Buchhalter, I. et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. International journal of cancer 144, 848–858; 10.1002/ijc.31878 (2019).
5 Heeke, A. L. et al. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO precision oncology 2018; 10.1200/PO.17.00286 (2018).
6 Nguyen, L., W M Martens, J., van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nature communications 11, 5584; 10.1038/s41467-020-19406-4 (2020).
Weitere Informationen
Webinar: Entdecken Sie die Möglichkeiten der modernen Tumordiagnostik
Genetische Tumordiagnostik kann Leben retten
Hinweis: Die Inhalte sind nur in englischer Sprache verfügbar.
Downloads
Kontaktieren Sie uns
Sie haben noch eine Frage oder Interesse an unserem Service?
Diagnostik-Support
Wir unterstützen Sie auf Wunsch bei der Auswahl der diagnostischen Strategie – für jede einzelne Patientin und jeden einzelnen Patienten.
