Von knapp 800.000 jährlichen Neugeborenen in Deutschland weisen ca. 7 % (~56.000 Betroffene) unterschiedliche Fehlbildungen auf. Die Ursachen und der Schweregrad solcher Fehlbildungen sind sehr vielseitig und umfassen äußere wie auch erbliche (genetische) Faktoren. Es wird davon ausgegangen, dass mindestens 20 % aller Fehlbildungen eine genetische Ursache haben, also eine Veränderung des Erbguts (DNA) zugrunde liegt. Viele Menschen tragen krankheitsverursachende Varianten in ihrem Erbgut, ohne es zu wissen und ohne selbst zu erkranken. Man spricht hierbei von „Anlagenträgerschaften“ für eine genetische Erkrankung. Anlagenträgerschaften sind von Mensch zu Mensch unterschiedlich. Sind beide Eltern gesund, aber Träger einer pathogenen Variante in einem Gen das ursächlich für rezessiv vererbte Erkrankungen ist, besteht ein 25-prozentiges Risiko, dass gemeinsame Nachkommen von der Erkrankung betroffen sind. Gemeinsame oder ähnliche Anlageträgerschaften können also zu einem Risiko für das Kind führen. Dies ist insbesondere bei gleicher ethnischer Herkunft des Paares, oder bei dem Verdacht auf eine genetische Erkrankung in einer der Familien der Fall. Im Falle einer gesicherten oder möglichen entfernten Verwandtschaft des Paares sind eine genetische Beratung und gegebenenfalls eine Vorsorgeuntersuchung ratsam.
Mit dem Family Planning Panel können Sie das genetische Risiko für Ihr Kind noch vor der Schwangerschaft ermitteln und so zu dessen Gesundheit beitragen.
Sie sind in Deutschland versichert? Unsere Kolleginnen und Kollegen vom Zentrum für Humangenetik Tübingen beraten Sie gerne!

Was wir Ihnen mit diesem Panel bieten
Unser Versprechen an Sie

Informationen zum Panel
Im Family Planning Panel werden insgesamt 1.943 Gene untersucht, die mit schweren frühkindlichen Erkrankungen assoziiert sind. Dabei werden nicht nur autosomal-rezessive Erkrankungen, sondern auch X-chromosomale Vererbung, genetisches Imprinting und dominant vererbte Erkrankungen bei Verdacht auf ein Keimbahnmosaik berücksichtigt. Hierbei werden sowohl in der Allgemeinbevölkerung häufige genetische Erkrankungen wie die cystische Fibrose (CF) oder die spinale Muskelatrophie (SMA) als auch sehr selten auftretende Syndrome gezielt untersucht. Für die Auswertung kombinieren wir die Daten beider Elternteile und ermitteln daraus das individuelle Risiko für Ihr Kind. Das Ergebnis der genetischen Untersuchung ermöglicht es Ihnen, eine fundierte Abschätzung der Risiken zu treffen und eröffnet die Möglichkeit einer vorgeburtlichen Diagnostik in der Schwangerschaft.
Leistungsumfang
- Umfangreiche Analyse von 1.943 Genen
- Die Analyse umfasst rezessive, X-chromosomale, Imprinting assoziierte sowie durch elterliche Mosaike entstandene frühkindliche Erkrankungen.
- Kombinationen von SNVs und CNVs werden berücksichtigt.
- Inkludiert ist eine Repeat-Analyse auf das Fragile-X-Syndrom (FMR1-Repeat) sowie eine Deletionsanalyse auf spinale Muskelatrophie (SMN1-MLPA).
- Die Daten beider Eltern werden zusammen analysiert und in einem leicht verständlichen Bericht zusammengefasst.
- Elterliche Varianten, die ein direktes Risiko für eine Erkrankung bei gemeinsamen Nachkommen bedingen, werden im Befund aufgeführt.
- Das Panel basiert auf der CeGaT ExomeXtra®-Anreicherung.
Zusätzlicher Service
- ACMG Gen-Panel – mehr Informationen
- Pharmakogenetik – mehr Informationen
Beispielbefund
Allgemeine Informationen
Material
- 1-2 ml EDTA-Blut
- Genomische DNA (1-2 µg)
- Einsendeformular mit Einverständniserklärung nach dem Gendiagnostikgesetz (GenDG)
Andere Probenmaterialquellen sind auf Anfrage möglich. Bitte beachten Sie, dass bei unzureichender Probenqualität die Analyse fehlschlagen kann. Wenn Sie mehr als eine Option an Proben haben, kontaktieren Sie uns bitte (diagnostic-support@cegat.de) und wir werden Ihnen bei der Auswahl der optimalen Probe helfen.
Dauer
- Dauer der Untersuchung: 3 – 4 Wochen
Methode
Die Anreicherung der kodierenden Regionen und der angrenzenden intronischen Regionen erfolgt durch eine In-Solution-Hybridisierungstechnologie. Die Auswahl der Zielregionen und das Design der Anreicherungs-Baits werden hausintern durchgeführt. Die Hochdurchsatzsequenzierung wird auf Illumina-Plattformen durchgeführt.
Die bioinformatische Verarbeitung der Daten erfolgt mit Hilfe eines unternehmenseigenen Rechenzentrums.
Im Anschluss an die Datenverarbeitung analysiert unser Team aus Wissenschaftlerinnen und Wissenschaftlern und Spezialistinnen und Spezialisten für Humangenetik die Daten und erstellt einen leicht verständlichen medizinischen Bericht.
Warum kann der Trägerstatus vor der Familienplanung überprüft werden?
Jeder gesunde Mensch trägt krankheitsverursachende genetische Veränderungen in sich, die in bestimmten Konstellationen zum Auftreten einer Erkrankung führen. Eine genetische Untersuchung mit der Partnerin, bzw. dem Partner, hilft bei der Abschätzung des Risikos für schwere genetische Erkrankungen bei gemeinsamen Nachkommen. In den folgenden Grafiken wird das Vererbungsmuster in einer Familienkonstellation erklärt:
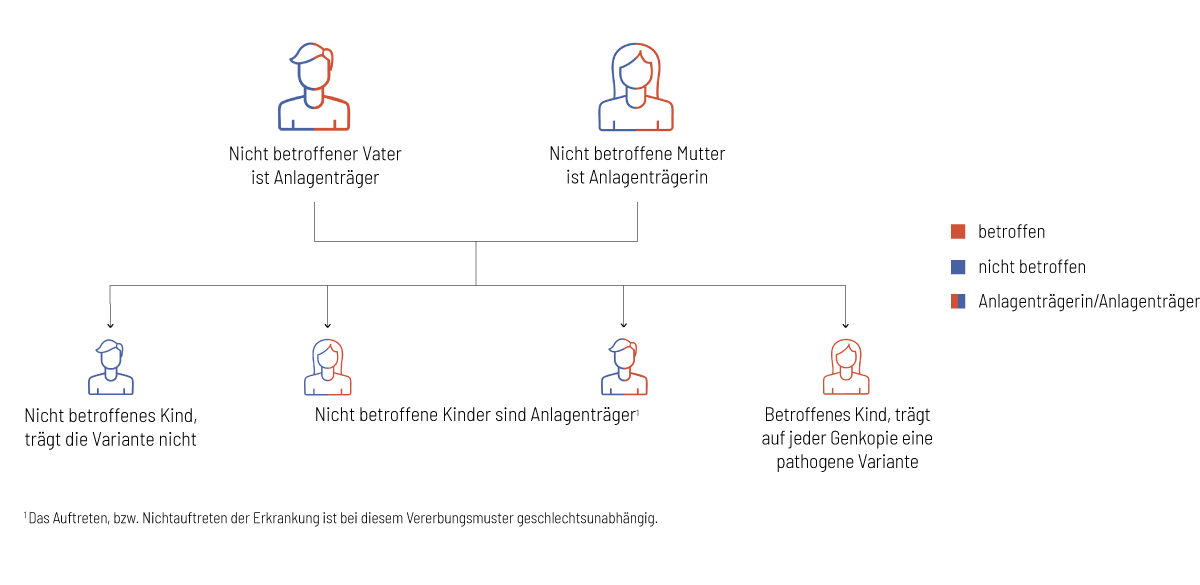
- Es besteht eine Wahrscheinlichkeit von 1:4 (25 %), dass das Kind keine veränderte Genkopie von den Eltern erbt und nicht von der genetischen Erkrankung betroffen ist.
- Es besteht eine Wahrscheinlichkeit von 1:2 (50 %), dass das Kind wie die Eltern Anlageträger, aber selbst nicht von der Erkrankung betroffen ist.
- Es besteht eine Wahrscheinlichkeit von 1:4 (25 %), dass das Kind von jedem Elternteil eine veränderte Genkopie vererbt bekommt und von der genetischen Erkrankung betroffen ist.
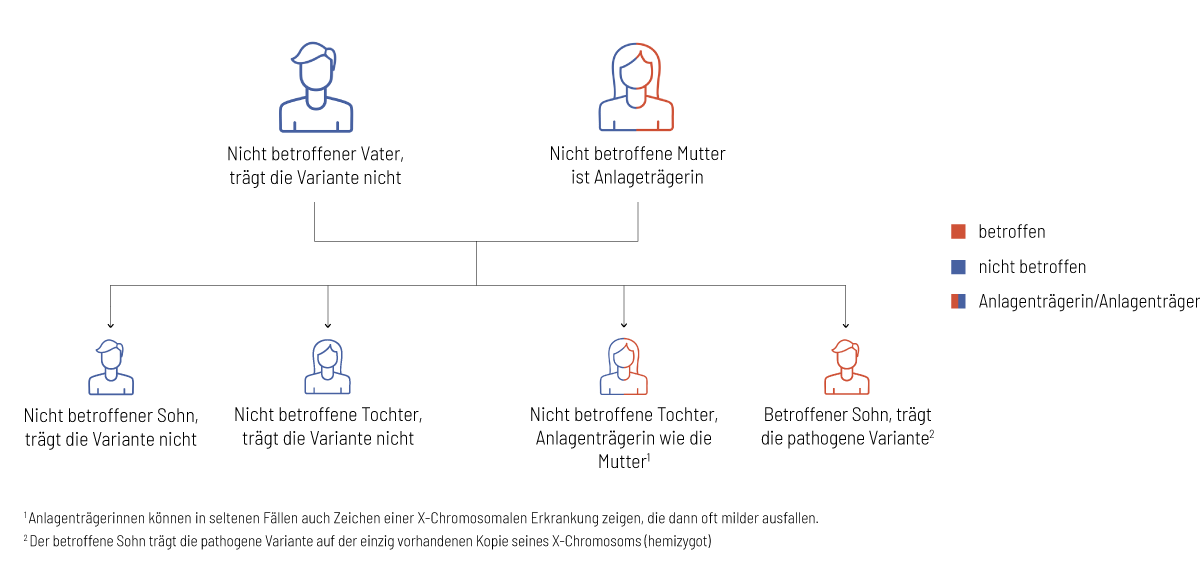
- Es besteht eine Wahrscheinlichkeit von 1:2 (50 %), dass Kinder die Variante nicht vererbt bekommen und nicht von der Erkrankung betroffen sind.
- Es besteht eine Wahrscheinlichkeit von 1:2 (50 %), dass Töchter Anlageträgerinnen sind (wie die Mutter), aber keine bzw. selten nur leichte Symptome aufweisen.
- Es besteht eine Wahrscheinlichkeit von 1:2 (50 %), dass Söhne die veränderte Genkopie erben und somit von der Erkrankung betroffen sind.
Genverzeichnis – Family Planning Panel
AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADSB, ACADVL, ACAN, ACAT1, ACD, ACE, ACO2, ACOX1, ACOX2, ACP5, ACSL4, ACTA1, ACTL6B, ACY1, ADA, ADA2, ADAM17, ADAM22, ADAMTS13, ADAMTS19, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY1, ADCY5, ADCY6, ADGRG1, ADGRG6, ADGRV1, ADK, ADPRS, ADSL, AFF2, AFG2A, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHI1, AIFM1, AIMP1, AIMP2, AIPL1, AIRE, AK2, AKR1D1, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALOXE3, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK3, ANKLE2, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP3D1, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AQP2, AR, ARFGEF2, ARG1, ARHGDIA, ARHGEF9, ARL13B, ARL3, ARL6, ARL6IP1, ARMC9, ARNT2, ARPC1B, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ATAD1, ATAD3A, ATCAY, ATIC, ATM, ATOH7, ATP13A2, ATP1A2, ATP2B3, ATP5F1D, ATP5MK, ATP6AP1, ATP6AP2, ATP6V0A2, ATP6V0A4, ATP6V1A, ATP6V1B1, ATP6V1E1, ATP7A, ATP7B, ATP8A2, ATP8B1, ATPAF2, ATR, ATRX, AUH, AVIL, B3GALNT2, B3GALT6, B3GAT3, B3GLCT, B4GALNT1, B4GALT1, B4GALT7, B4GAT1, B9D1, B9D2, BANF1, BBIP1, BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, BCAP31, BCKDHA, BCKDHB, BCKDK, BCOR, BCS1L, BGN, BHLHA9, BIN1, BLM, BLNK, BLTP1, BMP1, BMP2, BMPER, BMPR1B, BOLA3, BPNT2, BRAT1, BRCA1, BRCA2, BRF1, BRWD3, BSCL2, BSND, BTD, BTK, BUB1B, C12ORF57, C19ORF12, C1QBP, C2CD3, C2ORF69, CA2, CA5A, CA8, CABP2, CACNA1D, CAD, CAMK2A, CANT1, CAPN3, CARD11, CARMIL2, CARS2, CASK, CASQ2, CASR, CAV1, CAVIN1, CBS, CC2D1A, CC2D2A, CCBE1, CCDC103, CCDC115, CCDC22, CCDC39, CCDC40, CCDC47, CCDC65, CCDC8, CCDC88A, CCDC88C, CCN6, CCNO, CCNQ, CCT5, CD19, CD247, CD27, CD3D, CD3E, CD3G, CD40, CD40LG, CD55, CD70, CD79A, CD79B, CDC14A, CDC45, CDH11, CDH2, CDH23, CDH3, CDIN1, CDK10, CDK5RAP2, CDKL5, CDSN, CDT1, CENPF, CENPJ, CEP104, CEP120, CEP135, CEP152, CEP164, CEP290, CEP41, CEP55, CEP57, CEP63, CEP78, CEP83, CERS1, CERS3, CFAP298, CFAP300, CFAP410, CFAP418, CFL2, CFP, CFTR, CHAT, CHKB, CHM, CHMP1A, CHRDL1, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CHST14, CHST3, CHSY1, CHUK, CIB2, CIITA, CILK1, CISD2, CIT, CKAP2L, CLCN1, CLCN2, CLCN4, CLCN5, CLCN7, CLCNKB, CLDN1, CLDN10, CLDN14, CLDN16, CLDN19, CLIC5, CLMP, CLN3, CLN5, CLN6, CLN8, CLP1, CLPB, CLPP, CLRN1, CNKSR2, CNNM2, CNPY3, CNTNAP1, CNTNAP2, COA6, COA8, COASY, COCH, COG1, COG2, COG4, COG5, COG6, COG7, COL11A1, COL11A2, COL13A1, COL17A1, COL18A1, COL1A2, COL27A1, COL3A1, COL4A3, COL4A4, COL4A5, COL6A1, COL6A2, COL6A3, COL7A1, COL9A2, COLEC10, COLEC11, COLQ, COQ2, COQ4, COQ6, COQ7, COQ8A, COQ8B, COQ9, CORO1A, COX10, COX14, COX15, COX20, COX6A2, COX6B1, COX7B, COX8A, CPLANE1, CPLX1, CPS1, CPT1A, CPT2, CRADD, CRB1, CRB2, CRBN, CREB3L1, CRIPT, CRLF1, CRPPA, CRTAP, CRYAA, CRYAB, CSF1R, CSF2RB, CSF3R, CSPP1, CSTA, CSTB, CTC1, CTDP1, CTNNA2, CTNS, CTPS1, CTSA, CTSD, CTSK, CTU2, CUL4B, CUL7, CWC27, CWF19L1, CYB5R3, CYBA, CYBB, CYC1, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP24A1, CYP27A1, CYP27B1, CYP2R1, CYP2U1, CYP4F22, CYP7B1, D2HGDH, DAG1, DARS1, DARS2, DBT, DCAF17, DCDC2, DCHS1, DCLRE1C, DCX, DDB2, DDC, DDHD1, DDHD2, DDR2, DDX11, DDX3X, DDX59, DEAF1, DEGS1, DENND5A, DGAT1, DGKE, DGUOK, DHCR24, DHCR7, DHDDS, DHH, DHODH, DHTKD1, DHX37, DIAPH1, DIS3L2, DKC1, DLAT, DLD, DLG3, DLL3, DLX5, DMD, DMP1, DMXL2, DNA2, DNAAF11, DNAAF3, DNAAF4, DNAAF5, DNAAF6, DNAH11, DNAH5, DNAH9, DNAJC12, DNAJC19, DNAJC21, DNAJC3, DNAJC6, DNM1L, DNM2, DNMT3B, DOCK2, DOCK6, DOCK7, DOCK8, DOK7, DOLK, DONSON, DPAGT1, DPH1, DPM1, DPM2, DPYD, DRC1, DSE, DSG1, DSP, DST, DSTYK, DUOX2, DUOXA2, DYM, DYNC2H1, DYNC2I1, DYNC2I2, DYNC2LI1, DYSF, EARS2, EBP, ECEL1, ECHS1, EDA, EDAR, EDARADD, EDN3, EDNRB, EFEMP2, EFL1, EFNB1, EGR2, EIF2AK3, EIF2AK4, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S3, EIF4A3, ELAC2, ELMO2, ELMOD3, ELOVL4, ELP1, ELP2, EMC1, EMC10, EMD, EMG1, EML1, ENPP1, ENTPD1, EOGT, EPCAM, EPG5, EPM2A, EPRS1, EPS8, EPS8L2, ERAL1, ERBB3, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERCC6, ERCC6L2, ERCC8, ERLIN1, ERLIN2, ESCO2, ESPN, ESRRB, ETFA, ETFB, ETFDH, ETHE1, EVC, EVC2, EXOC3L2, EXOSC3, EXOSC8, EXOSC9, EXPH5, EXT2, EXTL3, F10, F13A1, F2, F7, F8, F9, FA2H, FADD, FAH, FAM149B1, FAM20A, FAM20C, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FAR1, FARS2, FASTKD2, FAT4, FBLN5, FBP1, FBXL4, FBXO7, FCSK, FERMT3, FEZF1, FGA, FGB, FGD1, FGD4, FGF3, FGFR3, FGG, FH, FHL1, FIG4, FITM2, FKBP10, FKBP14, FKRP, FKTN, FLAD1, FLNA, FLNB, FLVCR1, FLVCR2, FOLR1, FOXE1, FOXE3, FOXL2, FOXN1, FOXP3, FOXRED1, FRAS1, FREM1, FREM2, FRMPD4, FRRS1L, FSHB, FTCD, FTL, FTO, FTSJ1, FUCA1, FUT8, FXN, G6PC1, G6PC3, GAA, GAD1, GALC, GALE, GALK1, GALNS, GALT, GAMT, GAN, GAS8, GATA1, GATM, GBA1, GBA2, GBE1, GCDH, GCH1, GCK, GCSH, GDAP1, GDF1, GDF5, GDF6, GDI1, GEMIN4, GFER, GFM1, GFM2, GFPT1, GHR, GIPC3, GJA1, GJB2, GJB3, GJB6, GJC2, GK, GLA, GLB1, GLDC, GLDN, GLE1, GLIS3, GLRX5, GLS, GLUL, GLYCTK, GM2A, GMPPA, GMPPB, GNB5, GNPAT, GNPTAB, GNPTG, GNRH1, GNRHR, GNS, GOLGA2, GORAB, GOSR2, GOT2, GPAA1, GPC3, GPC6, GPHN, GPSM2, GPT2, GPX4, GRHL2, GRHPR, GRIA3, GRID2, GRIK2, GRIN1, GRIP1, GRM1, GRM7, GRXCR1, GSS, GTF2H5, GTPBP3, GUCY1A1, GUCY2C, GUF1, GUSB, GYS1, GYS2, GZF1, HACD1, HACE1, HADH, HADHA, HADHB, HAMP, HARS1, HARS2, HAX1, HBB, HCCS, HCFC1, HDAC8, HEPACAM, HERC1, HERC2, HES7, HESX1, HEXA, HEXB, HGF, HGSNAT, HIBCH, HIKESHI, HINT1, HJV, HK1, HLCS, HMGCL, HMGCS2, HMX1, HNRNPH2, HOGA1, HOXA1, HOXC13, HPD, HPDL, HPGD, HPRT1, HPS1, HPSE2, HSD11B2, HSD17B10, HSD17B3, HSD17B4, HSD3B2, HSD3B7, HSPA9, HSPD1, HSPG2, HTRA2, HUWE1, HYAL1, HYCC1, HYLS1, IARS1, IARS2, IBA57, ICOS, IDS, IDUA, IER3IP1, IFIH1, IFNGR1, IFNGR2, IFT122, IFT140, IFT172, IFT27, IFT43, IFT52, IFT56, IFT74, IFT80, IFT81, IGBP1, IGF1, IGF1R, IGFBP7, IGHMBP2, IGSF1, IHH, IKBKB, IL10RA, IL11RA, IL12RB1, IL1RAPL1, IL1RN, IL21R, IL2RA, IL2RB, IL2RG, IL7R, ILDR1, INPP5E, INPP5K, INPPL1, INS, INSR, INTU, INVS, IPO8, IQCB1, IQSEC1, IQSEC2, IRAK4, IRF8, IRX5, ISCA1, ISCA2, ITCH, ITGA3, ITGA6, ITGA7, ITGA8, ITGB4, ITK, ITPA, ITPR1, IVD, JAGN1, JAK3, JAM2, JAM3, JUP, KARS1, KATNB1, KATNIP, KCNE1, KCNJ1, KCNJ10, KCNJ11, KCNMA1, KCNQ1, KCTD7, KDELR2, KDM5B, KDM5C, KDM6A, KIAA0586, KIAA0753, KIDINS220, KIF14, KIF1A, KIF1C, KIF7, KIFBP, KISS1R, KLHL15, KLHL40, KLHL41, KLHL7, KNL1, KPTN, KRT10, KRT14, KRT18, KRT5, KRT8, KY, L1CAM, L2HGDH, LAGE3, LAMA1, LAMA2, LAMA3, LAMB1, LAMB2, LAMB3, LAMC2, LAMC3, LAMP2, LARGE1, LARP7, LARS2, LAS1L, LAT, LBR, LDHA, LDLR, LFNG, LGI4, LHB, LHFPL5, LHX3, LIAS, LIFR, LIG4, LIMS2, LINS1, LIPA, LIPT1, LMBR1, LMBRD1, LMNA, LMOD3, LNPK, LONP1, LOXHD1, LPIN1, LPIN2, LPL, LRBA, LRP2, LRP4, LRP5, LRPPRC, LRRC56, LRTOMT, LTBP2, LTBP3, LTBP4, LYRM4, LYRM7, LYST, LZTFL1, LZTR1, MAB21L2, MAG, MAGI2, MAGT1, MALT1, MAMLD1, MAN1B1, MAN2B1, MANBA, MAOA, MAP3K20, MAPKBP1, MARS1, MARVELD2, MASP1, MAT1A, MATN3, MBOAT7, MBTPS2, MC2R, MCCC1, MCCC2, MCEE, MCM4, MCOLN1, MCPH1, MDH2, MECP2, MECR, MED12, MED17, MED23, MED25, MEGF10, MEGF8, MEOX1, MESD, MESP2, MET, METTL23, METTL5, MFN2, MFRP, MFSD2A, MFSD8, MGAT2, MGME1, MGP, MICOS13, MICU1, MID1, MIPEP, MITF, MKKS, MKS1, MLC1, MLPH, MLYCD, MMAA, MMAB, MMACHC, MMADHC, MMP13, MMP2, MMP21, MMUT, MOCS1, MOCS2, MOGS, MPDU1, MPDZ, MPI, MPL, MPLKIP, MPV17, MPZ, MPZL2, MRE11, MRPL3, MRPL44, MRPS14, MRPS16, MRPS2, MRPS22, MRPS34, MSL3, MSMO1, MSN, MSRB3, MSTO1, MTFMT, MTHFD1, MTHFR, MTM1, MTMR2, MTO1, MTR, MTRFR, MTRR, MTTP, MUSK, MUTYH, MVK, MYBPC1, MYBPC3, MYD88, MYH11, MYH3, MYH7, MYL3, MYMK, MYO15A, MYO18B, MYO3A, MYO5A, MYO5B, MYO6, MYO7A, MYO9A, MYOD1, MYPN, MYSM1, NAA10, NADSYN1, NAGA, NAGLU, NAGS, NALCN, NANS, NARS1, NARS2, NAXD, NAXE, NBAS, NBN, NCAPD3, NCF1, NCF2, NCF4, NCKAP1L, NDE1, NDP, NDRG1, NDST1, NDUFA1, NDUFA10, NDUFA11, NDUFA12, NDUFA13, NDUFA2, NDUFA6, NDUFA9, NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB3, NDUFB8, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NEB, NECAP1, NECTIN1, NECTIN4, NEK1, NEK8, NEK9, NEMF, NEU1, NEUROG3, NEXMIF, NFASC, NFU1, NGF, NGLY1, NHEJ1, NHLRC1, NHP2, NHS, NIPAL4, NKAP, NKX3-2, NKX6-2, NMNAT1, NNT, NODAL, NONO, NOP10, NPC1, NPC2, NPHP1, NPHP3, NPHP4, NPHS1, NPHS2, NPR2, NR0B1, NR1H4, NRROS, NRXN1, NSDHL, NSMCE2, NSMCE3, NSUN2, NT5C2, NT5C3A, NTNG2, NTRK1, NUBPL, NUDT2, NUP107, NUP133, NUP188, NUP62, NUP88, NUP93, NYX, OBSL1, OCLN, OCRL, ODAD1, ODAD2, OFD1, OGDH, OPA1, OPA3, OPHN1, ORAI1, ORC1, ORC4, ORC6, OSGEP, OSTM1, OTC, OTOA, OTOF, OTOG, OTOGL, OTUD5, OTUD6B, OTULIN, OXCT1, OXR1, P3H1, PAH, PAK3, PAM16, PANK2, PAPSS2, PARN, PARS2, PAX3, PC, PCBD1, PCCA, PCCB, PCDH12, PCDH15, PCDH19, PCK1, PCNT, PCSK1, PCYT1A, PCYT2, PDE10A, PDE6D, PDE6G, PDHA1, PDHB, PDHX, PDP1, PDSS1, PDSS2, PDZD7, PEPD, PERCC1, PET100, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PFKM, PGAP1, PGAP2, PGAP3, PGK1, PGM1, PGM3, PHEX, PHF6, PHF8, PHGDH, PHKG2, PHYH, PI4KA, PIBF1, PIEZO1, PIEZO2, PIGA, PIGB, PIGG, PIGK, PIGL, PIGN, PIGO, PIGP, PIGQ, PIGS, PIGT, PIGV, PIGY, PIK3CD, PIK3R1, PIP5K1C, PISD, PITX3, PJVK, PKD1L1, PKHD1, PKLR, PLA2G6, PLAA, PLCB1, PLCB4, PLCE1, PLEC, PLEKHG2, PLEKHG5, PLG, PLK4, PLOD1, PLOD2, PLOD3, PLP1, PLPBP, PLS3, PLVAP, PMM2, PMP22, PMPCA, PMPCB, PNKP, PNP, PNPLA1, PNPLA6, PNPLA8, PNPO, PNPT1, POC1A, POC1B, POLA1, POLE, POLG, POLG2, POLR1C, POLR1D, POLR3A, POLR3B, POMC, POMGNT1, POMGNT2, POMK, POMP, POMT1, POMT2, POP1, POR, PORCN, POU1F1, POU3F4, PPA2, PPIB, PPIP5K2, PPP1R15B, PPP1R21, PPT1, PQBP1, PRDM12, PRDM5, PRDX1, PREPL, PRF1, PRG4, PRICKLE1, PRKCD, PRKDC, PRKRA, PRMT7, PROC, PRODH, PROP1, PROS1, PRPS1, PRRX1, PRSS12, PRSS56, PRUNE1, PRX, PSAP, PSAT1, PSMB8, PSPH, PTCHD1, PTF1A, PTH1R, PTPN14, PTPN23, PTPRC, PTPRQ, PTRH2, PTS, PUS1, PUS7, PXDN, PYCR1, PYCR2, PYGL, PYGM, PYROXD1, QARS1, QDPR, RAB18, RAB23, RAB27A, RAB33B, RAB39B, RAB3GAP1, RAB3GAP2, RAC2, RAD21, RAD50, RAD51C, RAG1, RAG2, RALGAPA1, RAPSN, RARB, RARS1, RARS2, RAX, RBBP8, RBCK1, RBM10, RBM8A, RDH11, RDX, RECQL4, RELN, REN, RETREG1, RFT1, RFX5, RFX6, RFXANK, RFXAP, RIC1, RIMS2, RIN2, RINT1, RIPK1, RIPK4, RIPOR2, RLIM, RMND1, RMRP, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2, RNF113A, RNF13, RNF168, RNU4ATAC, ROBO3, ROGDI, ROR1, ROR2, RPE65, RPGRIP1, RPGRIP1L, RPIA, RPL10, RPS6KA3, RRM2B, RSPH1, RSPH3, RSPO2, RSPO4, RSPRY1, RTEL1, RTN4IP1, RTTN, RUSC2, RXYLT1, RYR1, S1PR2, SACS, SAMD9, SAMHD1, SAR1B, SARS2, SASS6, SBDS, SBF1, SBF2, SC5D, SCAPER, SCARB2, SCARF2, SCN1B, SCN4A, SCN9A, SCNN1A, SCNN1B, SCNN1G, SCO1, SCO2, SCYL1, SCYL2, SDCCAG8, SDHA, SDHAF1, SDHD, SEC23A, SEC23B, SEC24D, SELENOI, SELENON, SEPSECS, SERAC1, SERPINB6, SERPINF1, SERPINH1, SETX, SFTPB, SFXN4, SGCA, SGCB, SGCD, SGCG, SGO1, SGPL1, SGSH, SH2D1A, SH3PXD2B, SH3TC2, SHOX, SIL1, SKIC2, SKIC3, SLC10A7, SLC12A1, SLC12A3, SLC12A5, SLC12A6, SLC13A5, SLC16A1, SLC16A2, SLC17A5, SLC18A3, SLC19A2, SLC19A3, SLC1A4, SLC22A5, SLC25A1, SLC25A12, SLC25A13, SLC25A15, SLC25A19, SLC25A20, SLC25A22, SLC25A26, SLC25A3, SLC25A38, SLC25A4, SLC25A42, SLC25A46, SLC26A2, SLC26A3, SLC26A4, SLC26A5, SLC27A4, SLC29A3, SLC2A1, SLC2A10, SLC2A2, SLC30A10, SLC33A1, SLC34A1, SLC34A3, SLC35A1, SLC35A2, SLC35A3, SLC35C1, SLC35D1, SLC37A4, SLC39A13, SLC39A14, SLC39A4, SLC39A8, SLC3A1, SLC46A1, SLC4A1, SLC4A4, SLC52A2, SLC52A3, SLC5A1, SLC5A5, SLC5A6, SLC5A7, SLC6A3, SLC6A5, SLC6A8, SLC6A9, SLC7A7, SLC9A1, SLC9A3, SLC9A6, SLX4, SMAD4, SMARCAL1, SMC1A, SMOC1, SMPD1, SMPD4, SMS, SNAP29, SNORD118, SNX10, SNX14, SOD1, SOST, SOX3, SP110, SP7, SPAG1, SPARC, SPART, SPEG, SPG11, SPINK5, SPINT2, SPR, SPTBN2, SPTBN4, SQSTM1, SRD5A2, SRD5A3, SSR4, ST14, ST3GAL3, ST3GAL5, STAC3, STAG2, STAMBP, STAR, STAT1, STAT2, STAT5B, STIL, STIM1, STN1, STRA6, STRADA, STS, STT3A, STUB1, STX11, STXBP2, SUCLA2, SUCLG1, SUFU, SUMF1, SUOX, SURF1, SVBP, SYN1, SYNE1, SYNE4, SYNJ1, SYP, SZT2, TAC3, TACO1, TACR3, TAF1, TAF13, TAF2, TAF6, TAFAZZIN, TALDO1, TANGO2, TAP1, TAPT1, TARS2, TASP1, TAT, TBC1D20, TBC1D23, TBC1D24, TBC1D8B, TBCD, TBCE, TBCK, TBX15, TBX19, TBX22, TBX4, TBXAS1, TCAP, TCF12, TCIRG1, TCN2, TCTN2, TCTN3, TDP2, TECPR2, TECTA, TELO2, TENM3, TENT5A, TERT, TF, TFR2, TGDS, TGFB1, TGM1, TH, THOC2, THOC6, TIMM50, TIMM8A, TIMMDC1, TJP2, TK2, TMC1, TMCO1, TMEM107, TMEM126A, TMEM126B, TMEM132E, TMEM138, TMEM165, TMEM199, TMEM216, TMEM231, TMEM237, TMEM260, TMEM38B, TMEM67, TMEM70, TMEM94, TMIE, TMPRSS3, TMPRSS6, TMTC3, TMX2, TNFRSF11A, TNFRSF11B, TNFRSF13B, TNFSF11, TNNT1, TOE1, TOP3A, TP53RK, TPI1, TPK1, TPM3, TPP1, TPRKB, TPRN, TRAF3IP1, TRAIP, TRAK1, TRAPPC11, TRAPPC12, TRAPPC2, TRAPPC4, TRAPPC9, TRDN, TREX1, TRIM2, TRIM32, TRIM37, TRIOBP, TRIP11, TRIP13, TRIP4, TRIT1, TRMT1, TRMT10A, TRMT10C, TRMT5, TRMU, TRNT1, TRPM6, TRPV6, TSEN15, TSEN2, TSEN54, TSFM, TSHB, TSHR, TSPAN7, TSPEAR, TSPYL1, TTC19, TTC21B, TTC7A, TTC8, TTI2, TTN, TTPA, TUBGCP2, TUBGCP4, TUBGCP6, TUFM, TUSC3, TWIST2, TWNK, TXN2, TXNDC15, TXNL4A, TYK2, TYMP, TYR, TYRP1, UBA1, UBA5, UBE2A, UBE2T, UBE3B, UBR1, UCHL1, UFC1, UFM1, UGDH, UGP2, UGT1A1, UMPS, UNC13D, UNC80, UPF3B, UQCC2, UQCRB, UQCRC2, UQCRFS1, UQCRQ, UROC1, UROS, USB1, USH1C, USH1G, USH2A, USP18, USP53, USP9X, UVSSA, VAC14, VAMP1, VARS1, VARS2, VDR, VIPAS39, VLDLR, VMA21, VPS11, VPS13B, VPS13D, VPS33A, VPS33B, VPS37A, VPS41, VPS45, VPS51, VPS53, VRK1, VSX2, WARS2, WAS, WASHC5, WBP2, WDPCP, WDR19, WDR35, WDR4, WDR45, WDR45B, WDR62, WDR73, WDR81, WFS1, WHRN, WNK1, WNT1, WNT10A, WNT10B, WNT2B, WNT3, WNT4, WNT7A, WRAP53, WRN, WWOX, XIAP, XPA, XPC, XRCC2, XRCC4, XYLT1, XYLT2, YARS2, YIF1B, ZAP70, ZBTB24, ZC3H14, ZC4H2, ZDHHC9, ZFYVE26, ZIC3, ZMPSTE24, ZNF335, ZNF711, ZNHIT3 (1937 Gene)
Berichtet werden krankheitsrelevante Veränderungen (SNV/CNV) in den oben gelisteten Genen (einem rezessiven/X-Chromosomalen Erbgang unterliegende Erkrankungen). Zusätzlich werden ggf. krankheitsverursachende Veränderungen (SNV/CNV) berichtet in weiteren Genen, die bekanntermaßen einem genetischen Imprinting unterliegen. Zudem ist ggf. das Berichten von Varianten möglich in weiteren Genen, die mit dominant vererbten Erkrankungen einhergehen, sofern diese bei einem der Ratsuchenden als genetisches Mosaik vorliegen. Berichtet werden ausschließlich Varianten in Genen, die im Rahmen der jeweils zugrunde liegenden Erbgänge beim geplanten Nachwuchs des ratsuchenden Paares potenziell zu schwerwiegenden Erkrankungen mit angenommenen Krankheitsausbruch im Kindesalter führen können. Einzelne Varianten eines Elter, die beim Nachwuchs zu einer Anlagenträgerschaft ohne erwartbaren Krankheitsausbruch führen können, werden nicht berichtet. Die Befundung beschränkt sich auf nach aktueller Datenlage gesichert krankheitsrelevante Varianten der ACMG Klassen 4(LP)/5(P). Einzelne ACMG Klasse 3(VUS) Varianten können in Einzelfällen nach ärztlichem Ermessen ggf. zusätzlich berücksichtigt werden.
Zusatzleistungen
HLA-Typisierung (HLA01)
HLA Allel-Status (HLA Klasse I (Gene A, B, C) und HLA Klasse II (Gene DPA1, DPB1, DQA1, DQB1, DRB1, DRB3, DRB4, DRB5))
ACMG-Gene
In seltenen Fällen können genetische Veränderungen nachgewiesen werden, die nicht im Zusammenhang mit dem Untersuchungsauftrag stehen (sog. Zusatzbefunde). Das Berichten solcher Zusatzbefunde beschränkt sich auf pathogene Veränderungen (ACMG Klassen 4 und 5) in ausgewählten Genen, für die eine Behandlungskonsequenz für die Patientin/den Patient oder die Familie besteht (orientiert an den aktuell gültigen Richtlinien des American College of Medical Genetics and Genomics).
Weitere Details zu den ACMG Genen und assoziierten Erkrankungen
Pharmakogenetik (PGX)
Die Pharmakogenetische Analyse detektiert genetische Veränderungen, die die Wirksamkeit von Medikamenten beeinflussen. Betreffen genetische Varianten Proteine, die für die Verstoffwechslung von Substanzen zuständig sind, kann deren Verträglichkeit und Wirksamkeit stark verändert sein. Zu diesen Arzneistoffen zählen unter anderem Antidepressiva, Schmerzmittel, Neuroleptika, Chemotherapeutika, AIDS-Medikamente, Thrombosemedikamente, Anästhetika, Betablocker oder Statine.
Die verringerte Aktivität eines spezifischen Enzyms kann bei der Standarddosierung zu einem erhöhten Medikamentenspiegel führen, der nicht selten mit unerwünschten Nebenwirkungen einhergeht. Bei Medikamenten, die erst durch die Verstoffwechslung aktiviert werden, kann der therapeutische Effekt ganz ausbleiben. Ebenso führt eine erhöhte Enzymaktivität, aufgrund der daraus resultierenden erhöhten Abbaugeschwindigkeit des Arzneistoffes, zu einer unzureichenden Wirksamkeit der Therapie.
Bei der Option „Pharmakogenetik“ werden bekannte Varianten in 21 Genen analysiert, die an der Verstoffwechselung von Arzneimitteln beteiligt sind. Bei Vorkommen bestimmter Genvarianten kann der behandelnde Arzt oder die behandelnde Ärztin die Therapie individuell anpassen. Mithilfe der pharmakogenetischen Analyse können gravierende Nebenwirkungen minimiert sowie ein Versagen der Therapie vermieden werden.
Details zu den Pharmakogenetik Genen und weitere Informationen
Häufig gestellte Fragen
Kontaktieren Sie uns
Sie haben noch eine Frage oder Interesse an unserem Service?
Diagnostik-Support
Wir unterstützen Sie auf Wunsch bei der Auswahl der diagnostischen Strategie – für jede einzelne Patientin und jeden einzelnen Patienten.
