Der erste Schritt für ein erfolgreiches Next-Generation-Sequencing-Projekt ist die Vorbereitung der Sequenzierlibrary. In diesem Schritt wird die Nukleinsäure für die Sequenzierung vorbereitet. In diesem Dokument möchten wir Ihnen zeigen, welche Schritte für eine erfolgreiche Vorbereitung der Nukleinsäuren notwendig sind, bevor die Proben sequenziert werden. Als Beispiel verwenden wir ein Projekt, in dem das ganze Genom sequenziert wird.
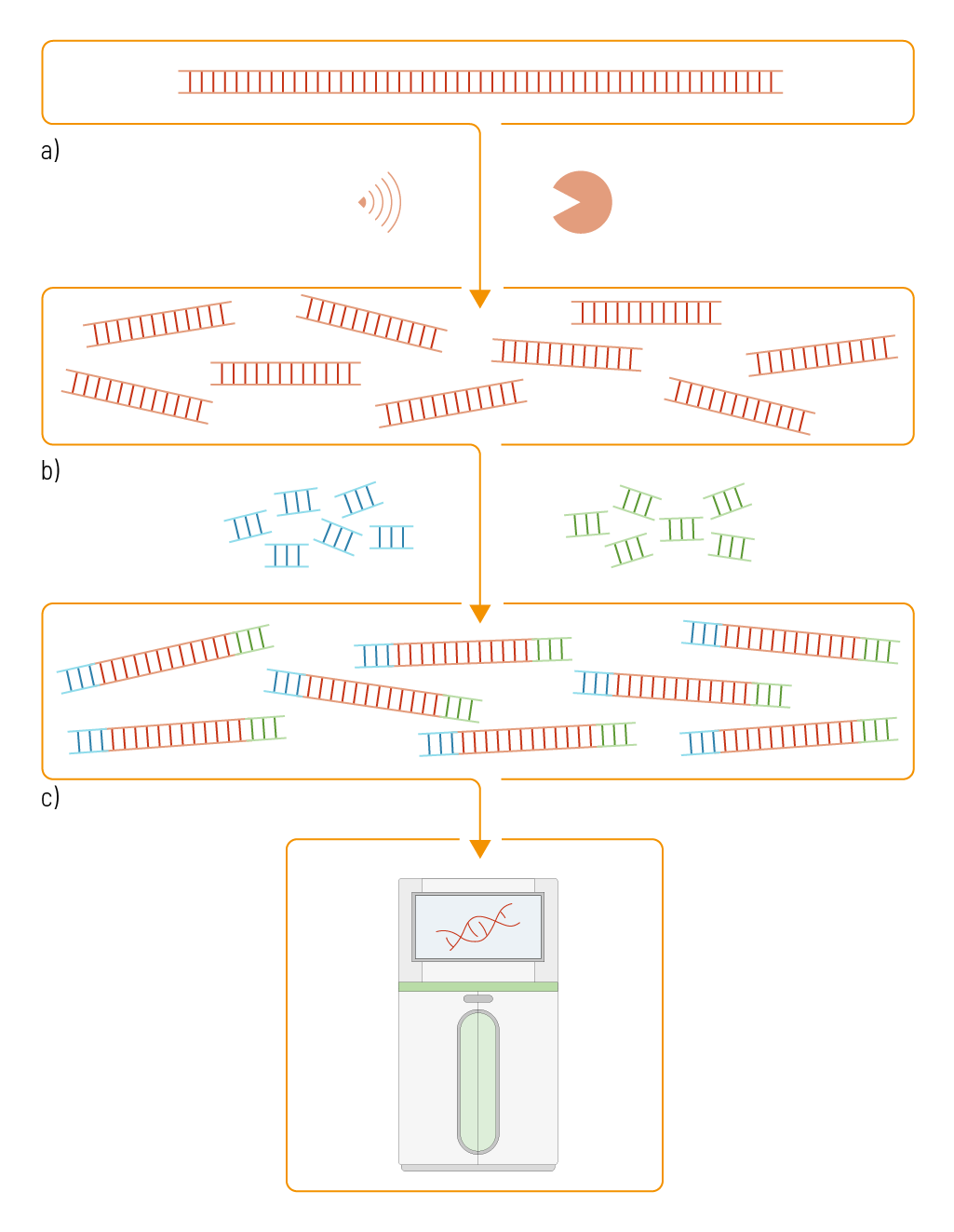
Abbildung 1 | Vorbereitungsprozess für die Sequenzierlibrary. Die gesamte Genomsequenz wird mit hochfrequenten Schallwellen oder Enzymen fragmentiert. An die entstehenden Nukleinsäure-Stücke werden Adapter hinzugefügt, welche Informationen für die Sequenzierung und den Ursprung der Probe enthalten. Die so entstandene Library kann dann sequenziert werden.
Die Sequenz des diploiden humanen Genoms beinhaltet im Durchschnitt ungefähr 6,32 Gigabasenpaare (Gbp) und ist mehr als 2 Meter lang. Um mit einer so langen Sequenz arbeiten zu können, wird sie erst zufällig in kürzere Stücke geteilt. Die Stücke haben dabei eine spezifische, vorher definierte Größe. Dieser Schritt wird Fragmentierung genannt (Abbildung 1a). Für die Fragmentierung gibt es verschiedene Möglichkeiten. Beim Ansatz der physikalischen Fragmentierung werden hochfrequente Schallwellen verwendet, um die DNA zu zertrennen. Eine andere Möglichkeit ist die Nutzung von Enzymen, die die DNA in kürzere Stücke schneiden. An jedes Ende dieser kurzen Nukleinsäure-Stücke werden Adapter hinzugefügt (Abbildung 1b). Adapter sind DNA-Sequenzen die Informationen für die Sequenzierung enthalten. Die Adapter enthalten zum einen eine Bindesequenz für die Flow Cell. Mit dieser bindet das kurze Nukleinsäure-Stück an die Flow Cell, wo dann die Sequenzierung stattfindet. Diese Bindesequenz für die Flow Cell ist Plattform-spezifisch. Die Adapter beinhalten auch eine Bindestelle für die Sequenzier-Primer, welche die Bindung des Primers, die Rekrutierung der Polymerase und die Verlängerung der Oligosynthese ermöglicht. Zusätzlich enthalten die Adapter einen sogenannten Index, um den Ursprung jeder Probe identifizieren zu können. Mit diesen Adaptern können die kurzen Nukleinsäure-Stücke vieler verschiedener Proben zusammen sequenziert werden, da jedes DNA-Stück anschließend wieder zu seiner ursprünglichen Probe zurücksortiert werden kann (Abbildung 1c).
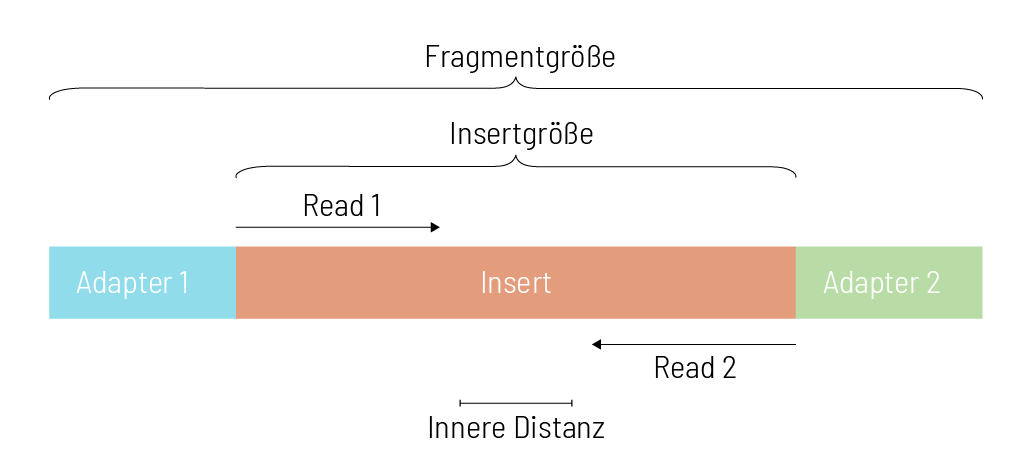
Diese kurzen Nukleinsäure-Stücke mit Adaptern werden Sequenzier-Fragmente genannt. Was ist aber nun die Fragmentgröße und was beinhaltet sie? Wie hängt die Fragmentgröße mit der Insertgröße zusammen?
Wir beginnen von innen: Das Stück DNA, das uns interessiert, ist das sogenannte Insert. In unserem Beispielprojekt mit der Genom-Sequenzierung sind die Inserts die Ergebnisse der Fragmentierung. Eine Verteilung von Inserts mit verschiedenen Längen entsteht dadurch, dass die zufällige Zerteilung während des Fragmentierungsprozesses nicht völlig regelmäßig geschieht. Die Länge der Inserts wird Insertgröße gennannt („insert size“). Auf Grundlage der Verteilung der Insertgrößen kann der Median der Insertgröße berechnet werden. Für unser Beispiel nehmen wir an, dass der Median der Insertgröße 318 bp beträgt.
Bei der Paired-End-Sequenzierung entsteht während des Sequenzierungsprozesses jeweils ein Read von jedem Ende des Inserts. Normalerweise ist die Insertgröße länger als die Summe der beiden Reads. Dies führt zu einem inneren Teil des Inserts, der nicht sequenziert wird. Die Länge dieses inneren Teils wird auch Innere Distanz genannt. In unserem Beispiel wurden die Fragmente im Paired-End-Sequenziermodus mit 150 bp sequenziert. Für beide Reads erhalten wir also 2 x 150 bp = 300 bp. Der Median der Insertgröße beträgt allerdings 318 bp, was zu einer inneren Distanz von 18 bp führt. Durch die Verwendung von kleineren Insertgrößen entsteht zwar keine innere Distanz, allerdings können Positionen in der Mitte doppelt sequenziert werden – ein Mal von jeder Seite des Inserts. Wenn die Insertgröße sogar kleiner als die Reandlänge ist, dann reicht das Read in die Adaptersequenz des anderen Endes hinein.
Das Fragment der Sequenzierung beinhaltet das Insert und die zwei Adapter an jedem Ende. Die Größe des Inserts und der beiden Adapter wird Fragmentgröße genannt.