The first step for a successful project using next-generation sequencing is the library preparation. In this step, the sample’s nucleic acid is prepared for the sequencing process. Within this work, we want to show which steps are necessary for the successful preparation of the nucleic acid before sequencing can be performed. Exemplarily, we are using a whole genome sequencing approach.
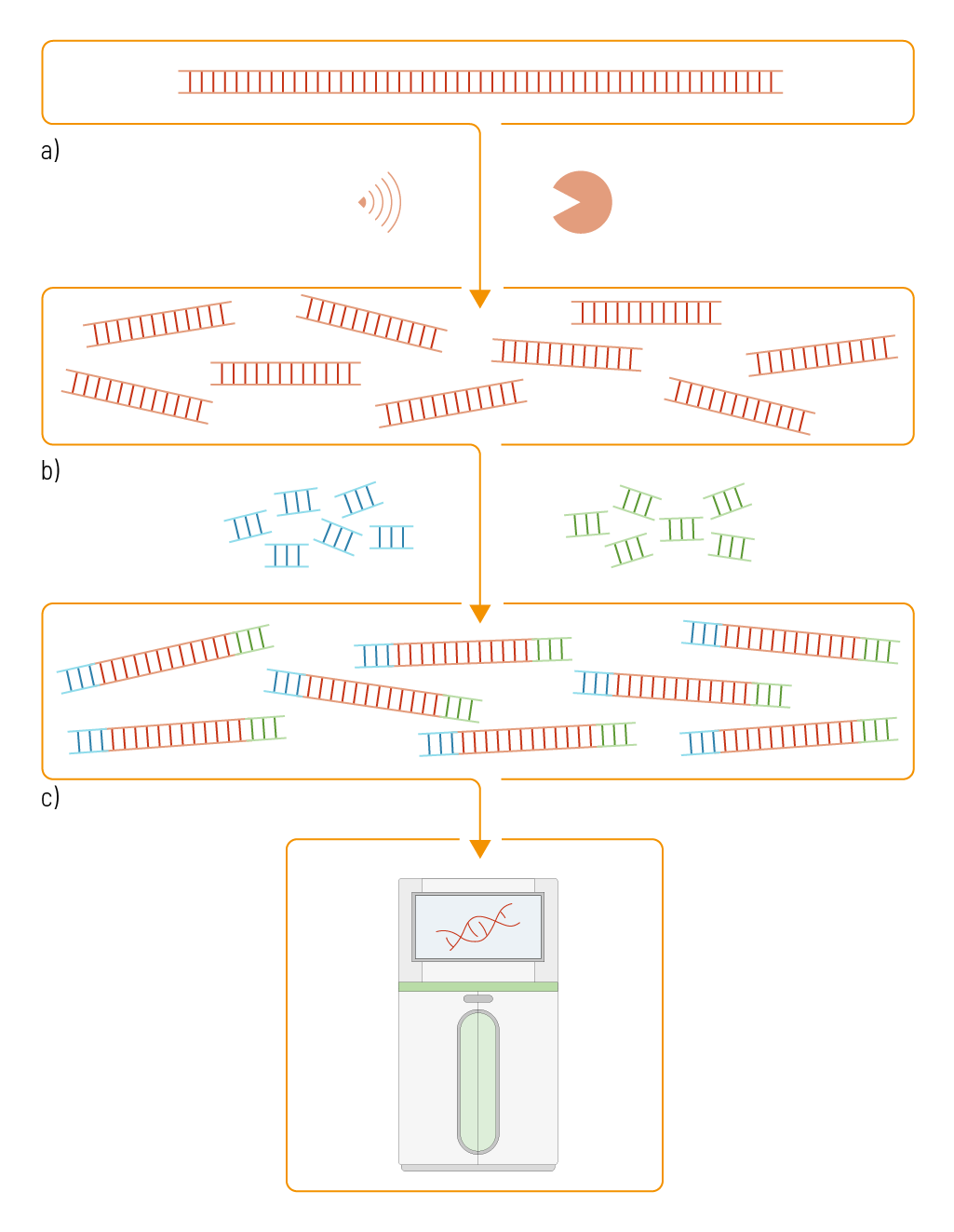
Figure 1 | Library preparation process. The complete genome sequence is fragmented with high-frequency sound waves or enzymes. To the resulting short nucleic acid pieces, adapters are added that contain the information required for sequencing and the sample’s origin. The resulting library can then be sequenced.
The sequence of the diploid human genome holds, on average, approximately 6.32 Gigabase pairs (Gbp) and is over 2 meters long. To work with such a long sequence, it first needs to be randomly cut into shorter pieces of a specific, predefined size. This step is called fragmentation (figure 1 a). For the fragmentation, different methods are available. In a physical fragmentation approach, high-frequency sound waves are used to shear the DNA. Another possibility is to use enzymes that cut the DNA into shorter pieces. To each end of these shorter nucleic acid pieces, adapters are added (figure 1 b). Adapters are DNA sequences that contain the information required for sequencing. These adapters contain a flow cell binding sequence with which they bind to the flow cell, where the actual sequencing occurs. This flow cell binding sequence is platform-specific. The adapters also contain a sequencing primer binding site, allowing the binding of the primer, recruiting the polymerase, and extending the oligo synthesis. Additionally, the adapters contain a so-called index to identify the sample‘s origin. With these adapters, short nucleic acid pieces of multiple samples can be sequenced together, as each piece can be sorted back to its initial sample afterward (figure 1 c).
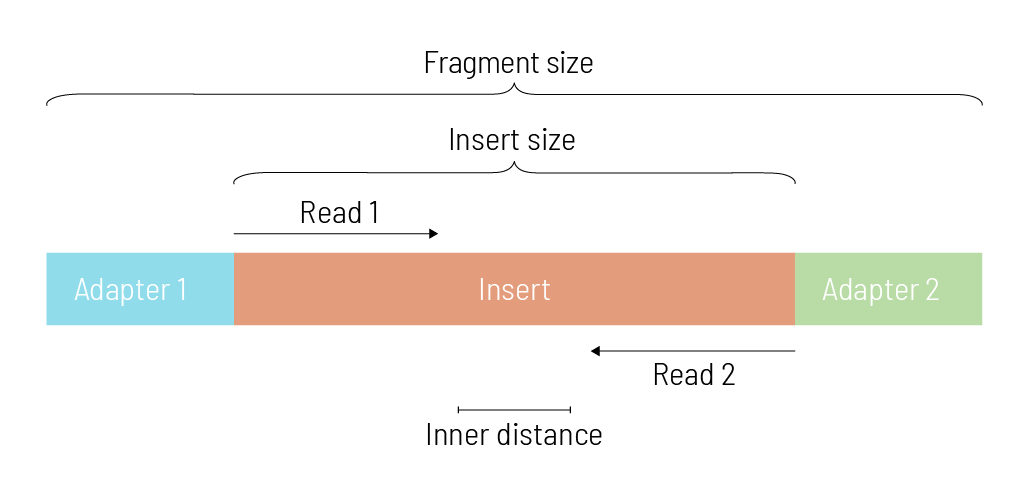
These short nucleic acid pieces with adapters are called sequencing fragments. But what exactly is the fragment size, and what does it contain? How is the fragment size connected to the insert size?
Let’s start from the inside: The piece of DNA of interest is the insert. In our genome example, the inserts are the result of the fragmentation. A distribution of inserts of different lengths is generated, as the random shearing during the fragmentation does not occur perfectly regularly. The length of these inserts is called the insert size. Based on the distribution of insert sizes, the median insert size can be calculated. Let’s assume that the median insert size in our example is 318 bp.
In paired-end sequencing, two reads from either end of the insert emerge during the sequencing process. Usually, the insert size is longer than the sum of the two reads. This results in an inner part of the insert that remains unsequenced. The length of this inner part is called the inner distance. In our example, the fragments were sequenced in the paired-end sequencing mode with 150 bp. For both reads, we get 2 x 150 bp = 300 bp. The median insert size, however, is 318 bp, which leaves a remaining median distance of 18 bp. With smaller insert sizes, no inner distance occurs, but positions in the middle might be sequenced twice – once from either side of the insert. If the insert size is smaller than the read length, the read reaches into the adapter sequence at the other end of the fragment.
The sequencing fragment is the insert plus the two adapters on either side. The size of the insert and the two adapters is called the fragment size.