Das Transkriptom umfasst alle RNA-Moleküle, die in einer Zelle oder einem Gewebetyp zu einer bestimmten Zeit vorhanden sind. Ihr Vorhandensein und ihre Häufigkeit entsprechen dem aktuellen Stoffwechselzustand der Zellen. Durch verschiedene interne und externe Einflüsse kann sich dieser Zustand verändern. Die Transkriptom-Sequenzierung (“transcriptome sequencing”) ist eine leistungsstarke Methode zum Nachweis und zur Quantifizierung von RNA-Molekülen.
Die Anwendungsbereiche und Ziele der Transkriptom-Sequenzierung sind vielfältig und umfassen
- die Analyse der unterschiedlichen Genexpressionsniveaus und
- den Nachweis von alternativem Spleißen und bisher unbekannten Transkripten.
Wir bieten verschiedene Transcriptome-Sequencing-Produkte an, die einen zuverlässigen und genauen Aufschluss über die RNA-Moleküle in einer Zelle geben.


CeGaT ist der beste Partner für Ihr Sequenzierprojekt
Unser Engagement für Sie
Schnelle Bearbeitung
Bearbeitungszeit
≤ 15 Werktage
Hohe Qualität
Höchste Genauigkeit bei allen Prozessen
Sichere Datenlieferung
Sichere Bereitstellung der sequenzierten Daten über hausinterne Server
Sichere Aufbewahrung
Sichere Proben- und Datenaufbewahrung nach Projektabschluss
Unser Service
Uns ist eine umfangreiche und erstklassige Projektbegleitung wichtig − von der Auswahl des passenden Produktes bis zur Auswertung der Daten. Jedes Projekt wird von einer engagierten Wissenschaftlerin oder einem engagierten Wissenschaftler begleitet, so dass Ihnen während des gesamten Projektverlaufs eine Ansprechpartnerin oder ein Ansprechpartner zur Seite steht.
Unser Service umfasst:
- Ausführliche Projektberatung
- Produktauswahl abgestimmt auf Ihr Projekt
- Umfangreiche bioinformatische Auswertung Ihrer Daten
- Abschließender und detaillierter Projektbericht mit Informationen zur Probenqualität, Sequenzierparametern, bioinformatischen Analysen und Ergebnissen
Profitieren Sie von unserem hervorragenden Support und unseren akkreditierten Arbeitsabläufen.

Unser Produktportfolio für Transcriptome Sequencing
Wir bieten Whole Transcriptome Sequencing (WTS), Coding Transcriptome Sequencing (CTS) und Flexible Solutions (TS) an, um eine Vielzahl von Forschungsfragen abzudecken. Wünschen Sie zusätzlich zu den beinhalteten Leistungen bioinformatische Analysen Ihrer Daten? Jedes unserer Produkte kann durch weitere Dienstleistungen ergänzt werden. Wir beraten Sie gerne.
CTS Classic | WTS Classic | WTS Classic Deep | TS Flex |
Spezies | Spezies | Spezies | Spezies |
RNA-Qualität & Input | RNA-Qualität & Input | RNA-Qualität & Input | RNA-Qualität & Input |
RNA Target | RNA Target Gesamt-RNA | RNA Target Gesamt-RNA | RNA Target mRNA, Gesamt-RNA |
Sequenzierplattform | Sequenzierplattform | Sequenzierplattform | Sequenzierplattform |
Output | Output | Output | Output |
Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen | Beinhaltete Leistungen |
Bioinformatik
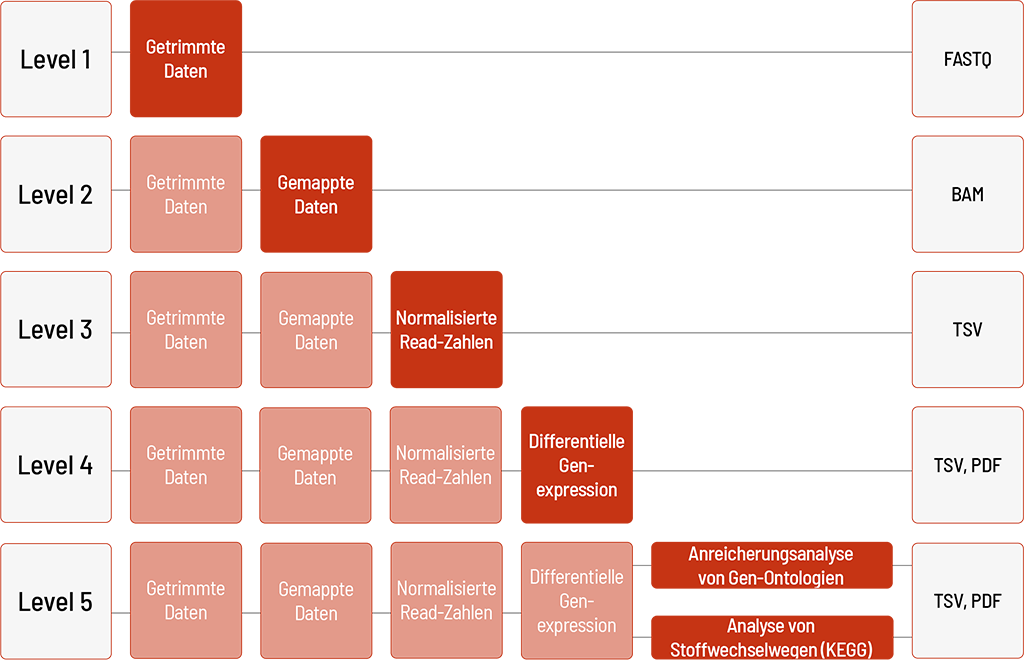
Die Rohdaten der Sequenzierung werden automatisch verarbeitet. Wir bieten verschiedene Level bioinformatischer Analysen an. Das Standardlevel ist Level 1. Mit steigendem Bioinformatiklevel werden mehr Daten geliefert. Alle höheren Level beinhalten dabei die Daten der vorherigen Level. Zusätzlich zu den Daten und unabhängig vom Analyselevel wird ein Projektbericht verfasst.
Level 1:
- Demultiplexing und Adapter-Trimming der Sequenzierdaten (FASTQ-Datei)
Level 2:
- Mapping der Sequenzierdaten (BAM-Datei)
Level 3:
- Normalisierung der Read Counts (TSV-Datei)
Level 4:
- Gruppenvergleich und differenzielle Genexpressionsanalyse (TSV-Datei)
- Visualisierung als Heatmap, MA-Plot und Vulcano-Plot (PDF-Datei)
⚠ Für den Gruppenvergleich sind mindestens drei Replikate pro Gruppe erforderlich.
Level 5 (eine der folgenden Möglichkeiten):
- Anreicherungsanalyse von Gen-Ontologien (GO terms) (TSV- und PDF-Datei) − für ausgewählte Organismen
- Analyse von Stoffwechselwegen (KEGG) (TSV- und PDF-Datei) − nur für menschliche Proben
Technische Information
Bei CeGaT wird die Paired-End-Sequenzierung (2 x 100 bp) mit den Sequenzierplattformen von Illumina durchgeführt. Wenn Sie andere Sequenzierparameter benötigen, lassen Sie es uns gerne wissen! Wir können Ihnen weitere Lösungen anbieten.
Weitere Informationen zu Transcriptome Sequencing
Transcriptome Sequencing wird auch als RNA-Sequenzierung (RNA Seq) bezeichnet. Bei dieser Methode werden die zu einer bestimmten Zeit vorhandenen RNA-Moleküle innerhalb einer Probe analysiert. So wird der Stoffwechselzustand einer Probe dargestellt und spiegelt den biologischen Zustand der Zelle zu diesem Zeitpunkt wider. Transcriptome Sequencing ermöglicht die Identifizierung alternativer Gene, gespleißter Transkripte und posttranskriptioneller Modifikationen. Zusätzlich können Genfusionen und Veränderungen der Genexpression im zeitlichen Verlauf, zum Beispiel während einer Therapie, oder zwischen verschiedenen Gruppen untersucht werden. Mit Transcriptome Sequencing können also Genfunktion und Genstruktur interpretiert werden. Darüber hinaus kann die Funktion von nicht-proteinkodierenden RNAs analysiert werden. Transcriptome Sequencing kann sowohl molekulare Mechanismen von Krankheiten und deren Entstehung als auch spezifische biologische Prozesse aufdecken. Außerdem können seltene oder unbekannte Transkripte sowie variable Spaltstellen (“cleavage sites”) und Einzelnukleotid-Polymorphismen der kodierenden Sequenz identifiziert werden.
RNA Seq hat gegenüber anderen Transcriptome-Sequencing-Technologien, wie z. B. Microarrays, einige Vorteile. Die Methode ist hochempfindlich und ermöglicht sowohl Nachweis als auch die Quantifizierung der Transkripte einer Zelle. Außerdem wird bei RNA Seq jedes einzelne Nukleotid eines Transkripts genau bestimmt. Im Unterschied zur Microarray-Technologie gibt es bei RNA Seq zum Beispiel keine Probleme mit Kreuzreaktionen oder Hintergrundrauschen von Fluoreszenzsignalen.
Innerhalb von RNA Seq können verschiedene Vorgehensweisen verwendet werden. Es ist entweder möglich, das gesamte Transkriptom (“whole transcriptome sequencing”) oder das kodierende Transkriptom (“coding transcriptome sequencing”) zu analysieren. Bei der Sequenzierung des gesamten Transkriptoms werden sowohl kodierende RNAs als auch nicht-kodierende RNAs erfasst. Im Gegensatz dazu zielt die Sequenzierung des kodierenden Transkriptoms auf die mRNA-Moleküle ab, um die Genexpression zu quantifizieren und die differentiellen Genexpression zu analysieren.
Je nach Versuchsaufbau oder Forschungsfrage kann die Gesamt-RNA oder die mRNA sequenziert werden. Die Gesamt-RNA umfasst sowohl die kodierende als auch nicht-kodierende RNA. Wir nennen dieses Produkt WTS, für Whole Transcriptome Sequencing. Die RNA-Isolierung kann aus Zellen, Blut oder sogar aus FFPE-eingebettetem Gewebe erfolgen und entweder von Ihnen oder von CeGaT durchgeführt werden. Die Gesamt-RNA enthält viele verschiedene RNA-Typen, wie z. B. mRNA, ribosomale RNA und nicht-kodierende RNA.
Nach der reversen Transkription, dem Hinzufügen von Adaptern und Barcodes und der PCR-Amplifikation wird die fertige Library auf einem unserer hochmodernen Sequenziergeräte sequenziert. Selbst schwierige Proben, wie z. B. fragmentierte RNA, lassen sich mit unseren Protokollen für Gesamt-RNA gut analysieren. Da die ribosomale RNA mehr als 80 % aller zellulären RNAs ausmacht, empfehlen wir die rRNA-Depletion. Wir können das Transkriptom von Säugetieren, Pflanzen, Viren und Bakterien analysieren und haben viele verschiedene Gesamt-RNA-Protokolle für viele verschiedene Anwendungen entwickelt. Die Transkriptom-Analyse ermöglicht einen umfassenden und genomweiten Überblick über alle Transkripte in einem Organismus sowie eine genomweite Expressionsanalyse. Es können Introns, Exons, aber auch Regionen dazwischen analysiert werden, um Aufschluss über Spleißmechanismen zu erhalten.
Die Sequenzierung der kodierenden RNA ist der gängigste Ansatz für die Transkriptom-Sequenzierung. Hierfür bieten wir das Produkt CTS, für Coding Transcriptome Sequencing, an. Die mRNA macht nur 1-5 % der gesamten RNA aus. Sie hat einen kodierenden Bereich, der am 5′- und 3′-Ende von zwei UTRs, den so genannten untranslatierten Bereichen (“untranslated regions”), flankiert wird. Diese tragen eine 5′-Cap-Struktur und eine 3′- Polyadenylierung, den so genannten Poly-A-Schwanz. Dieser Poly-A-Schwanz erhöht die Stabilität und die Translationseffizienz der mRNA. Die meisten Eukaryoten wie Säugetiere, Insekten, Pflanzen, Pilze und Fische tragen diesen Poly-A-Schwanz. In der mRNA von Bakterien ist er jedoch nicht vorhanden. Dieser Poly-A-Schwanz ist für die Herstellung der mRNA-Library unerlässlich. Nach der Isolierung der RNA aus Zellen oder Gewebe muss die mRNA mit Hilfe von Poly-T-Oligos gereinigt werden, die an Magnetkügelchen gekoppelt sind und an den Poly-A-Schwanz binden. Die endgültige Sequenzier-Library wird dann auf einem unserer modernen Sequenziergeräte analysiert.
Wir können das kodierende Transkriptom von Säugetieren, Pflanzen und anderen Eukaryoten analysieren. Die mRNA-Sequenzierung ist sehr empfindlich, vor allem, wenn man mRNA analysieren will, die nur in sehr geringen Mengen exprimiert wird. Eines der Hauptziele der mRNA-Sequenzierung ist die Quantifizierung der Genexpression und die Analyse der differentiellen Genexpression. Sie wird häufig verwendet, um die Genexpression zwischen verschiedenen Gruppen zu vergleichen, insbesondere bei Experimenten zu Arzneimitteltherapien oder medikamentösen Behandlungen von Krankheiten.
Downloads
Kontaktieren Sie uns
Sie haben noch Fragen oder Interesse an unserem Service? Treten Sie gern mit uns in Kontakt. Wir werden uns schnellstmöglich um Ihr Anliegen kümmern.
Starten Sie Ihr Projekt mit uns
Gerne beraten wir Sie zu unseren Sequenzierdienstleitungen und erarbeiten mit Ihnen gemeinsam die beste Lösung, die auf Ihre klinische Studie oder Forschungsprojekt abgestimmt ist.
Bitte geben Sie, falls möglich, folgende Probeninformationen an: Ausgangsmaterial, Anzahl der Proben, bevorzugte Option für die Vorbereitung der Library, bevorzugte Sequenziertiefe und gewünschte bioinformatische Analysestufe.
