Every cancer is as unique as the person it affects. Tumors can carry different genetic alterations, even within the same type of cancer. A tumor’s genetic profile plays a decisive role in determining which therapies are effective and which are not. That’s why comprehensive molecular genetic analysis is essential. It is the only way to identify relevant alterations and systematically integrate them into therapy planning. This approach enables patients to receive tailored treatment options while avoiding unnecessary interventions.
With CancerPrecision®, you receive a comprehensive molecular genetic tumor analysis that captures all essential information. The analysis is based on next-generation sequencing (NGS) and is suitable for all solid tumor entities. CancerPrecision® provides the most comprehensive basis for decision-making in personalized therapies, regardless of tumor stage or primary localization.
More than 700 genes, over 30 therapy-relevant gene fusions, and key biomarkers such as tumor mutational burden (TMB), microsatellite instability (MSI), homologous recombination deficiency (HRD), and potential oncogenic viral infections are analyzed. A crucial component of the CancerPrecision® analysis is the comparison of tumor and normal tissue, which allows for a reliable distinction between somatic (acquired) and germline-associated (hereditary) alterations. Only in this way can genetic findings be correctly classified and misinterpretations avoided. Due to its high sensitivity, CancerPrecision® enables the detection of subclones and low-frequency resistance mutations to certain drugs. An additional RNA-based fusion gene analysis, as well as a gene expression analysis, can be requested.
The results are compiled in a clearly structured medical report. You will receive a complete molecular genetic tumor profile, including possible treatment options, so that you can make informed and patient-specific treatment decisions with confidence.
Are you insured in Germany? Our colleagues at the Zentrum für Humangenetik Tübingen will gladly support you!

CancerPrecision® Is the First Choice for Tumor Characterization
* Based on a high-quality sample with at least 20% tumor content.
Our Promise to You

Service Details
- Tumor-normal tissue comparison for precise results — learn more
- Full sequencing and analysis of more than 700 tumor-associated genes and fusions in more than 30 genes — learn more
- High average sequencing of 500-1,000x coverage allows detection of therapy-relevant variants in subclones
- Sensitivity: > 97.6%*; Specificity: > 99,9%
- Analysis of tumor mutational burden (TMB), microsatellite instability (MSI), and viral infection (HPV/EBV/MCV/CMV) — essential biomarkers for immunotherapies — learn more
- Analysis of homologous recombination deficiency (HRD) — an essential biomarker for PARP inhibition — learn more
- Analysis of single nucleotide variants (SNVs), insertions and deletions (indels), translocations, and copy number variants (CNVs) — learn more
- Besides therapy-relevant somatic (tumor-specific) mutations also disease causing and therapy-relevant germline variants are reported
- Selected pharmacogenetically relevant germline variants necessary for drug dose adjustment in your patients
- A list of all eligible drugs, with EMA and/or FDA approval, for which corresponding biomarkers could be detected in the tumor — learn more
- Detection of mosaic variants: CHIP (Clonal Hematopoiesis of Indeterminate Potential)
Optional Service:
- Multiplex immunofluorescence staining of 9 markers (CD45, PD-L1, LAG3, p-mTOR, p-ERK, p16, p53, HER2, TROP2) on a single tissue section — learn more
- RNA-based fusion transcript analysis from tumor RNA analyzing > 200 genes (CancerFusionRx®) — learn more
- Transcriptome sequencing of tumor RNA to gain further insights on significant expression changes and their potential therapeutic relevance — learn more
* Based on a high-quality sample with 20 % tumor content for detecting a somatic heterozygous variant.
Sample Report
Our Standard Sample Requirements
Normal Tissue
- 1–2 ml EDTA blood (recommended sample type) or
- Genomic DNA (1–2 μg)
Tumor Tissue Options
Tumor content at least 20%
- FFPE tumor block (min. tissue size 5 x 5 x 5 mm) (recommended sample type)
- Unstained FFPE tumor tissue slides (min. 10 slices, tissue size 5 x 5 mm)
- for staining: 5 μm thickness
- for nucleic acid extraction (DNA/RNA): 10 μm thickness
- Isolated tumor DNA (> 200 ng)
- Fresh frozen tumor tissue
- 3 x 10 ml cfDNA tubes for liquid biopsy
Here you can find more information on how to ship your sample safely.
Further Sample Materials
Other sample material sources are possible on request. Please note: in case of insufficient sample quality or tumor content, the analysis might fail.
If you have more than one option of tumor samples, please get in touch with us (tumor@cegat.com), and we will assist you in choosing the optimal specimen for your patient.
For highest accuracy, we require tumor and normal tissue for our somatic tumor diagnostics panel.
This Is What Makes Our CancerPrecision® Service Special
CancerPrecision® goes far beyond a conventional panel diagnostic: we capture the full molecular genetic tumor profile – including all genetic alterations relevant to tumor behavior, therapy response, or potential resistance mechanisms.
To ensure you can navigate the analysis results as effectively as possible, the medical report is clearly structured: results are organized into main categories, therapy-relevant findings are highlighted in color, and all content is presented clearly and concisely. In this way, we support you in making informed and individually appropriate therapy decisions.
Tumor to Normal Tissue Comparison
The only accurate way to determine somatic variants
We always sequence DNA from both the tumor and normal tissue, usually blood. This is the only way to reliably determine whether a genetic alteration is tumor-specific or inherited. Making this distinction is crucial for accurately assessing genetic alterations and their relevance to potential therapeutic strategies.
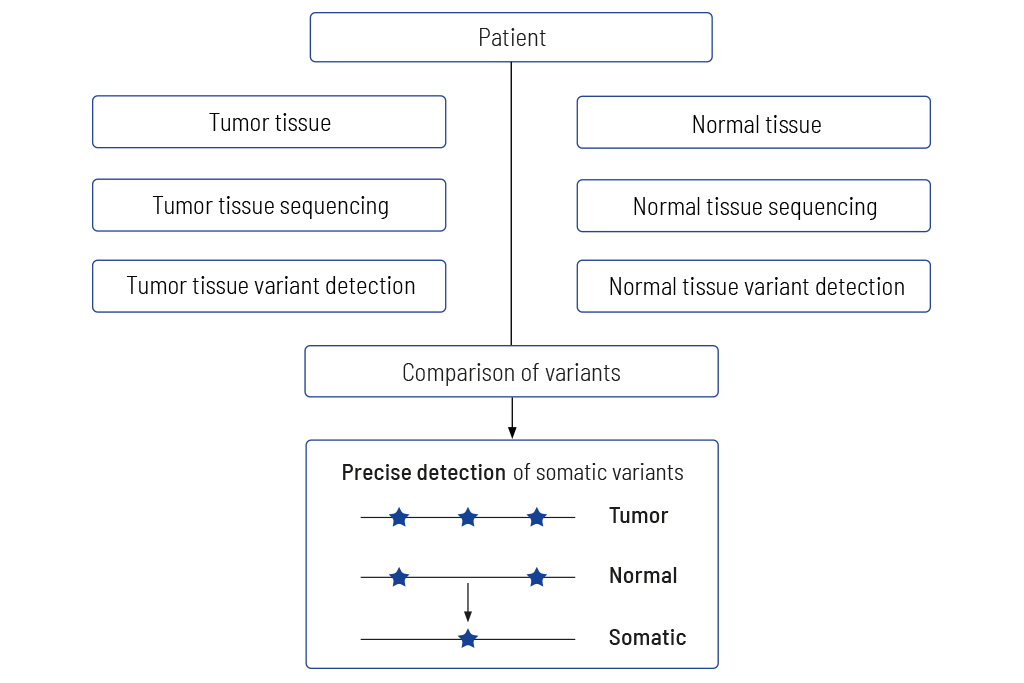
Without comparison to normal tissue, there is a risk of misclassifying genetic alterations – hereditary mutations could mistakenly be interpreted as tumor-specific changes, or vice versa. In addition, the presence of an inherited pathogenic alteration does not necessarily mean that it causes the tumor: only the comparison of tumor and normal tissue shows whether a pathogenic hereditary variant is a central driver of the tumor. This is usually confirmed by the detection of an additional tumor-specific genetic alteration (“second hit”) in the same gene that promotes tumor growth.
In up to 20% of all cases, cancer has a hereditary cause – an important diagnosis for patients and family members that should be discussed in genetic counseling. The detection of germline variants also determines eligibility for certain medications. Therefore, it is essential to classify genetic alterations precisely.
An accurate determination of molecular biomarkers such as TMB, MSI, or HRD is only possible by comparing tumor and normal tissue. Without this comparison, for example, the mutational burden may be overestimated, which can lead to the selection of unsuitable therapy options. Patients from population groups underrepresented in genomic databases particularly benefit from the tumor–normal tissue comparison, as it avoids reliance on unreliable bioinformatic estimation models to distinguish between hereditary and tumor-specific genetic alterations.
Genetic Variants with Potential Therapeutic Relevance
Guidance on potentially effective drugs
We analyze more than 700 genes and identify all variants with potential therapeutic relevance. For each relevant genetic alteration, the main section of the medical report provides detailed information on the type of alteration and the expected functional impact on protein structure or function. In addition, the potential therapeutic relevance is assessed. (A) This information forms the basis for discussion in a molecular tumor board (MTB).
In addition, the appendix of our medical report includes a therapy-related overview (B) for each identified alteration, provided approved drugs (FDA/EMA) are available. The individual tables focus on specific agents associated with the respective variant. With descending clinical relevance and selected approval restrictions are prechecked for applicability. Approved drugs based on the detected variants that fulfill the approval restrictions for the present entity is shown in the example.
This structured overview greatly facilitates the therapeutic classification of the genetic findings and provides a solid starting point for decisions on further treatment strategies.

Sample Report: Exemplary for the HRD and PIK3CA variant detected and the resulting therapeutic options in a EMA & FDA cancer patient. Top panel (A): An excerpt from Table 1 of the findings, listing variants with therapeutic relevance. Lower part (B): An excerpt of the drug listing. In addition to the drugs shown, other drugs are also described.
Pathway Illustration
For a detailed understanding of altered signaling
Tumors arise when the balance between cell growth and cell death is disrupted. During tumor development, both processes increasingly escape control, leading to abnormal cell growth.
Under normal conditions, cellular processes are precisely regulated by a complex network of signaling pathways. In tumors, however, mutations accumulate in genes that play central roles in these pathways. A single variant can already affect multiple signaling pathways – with direct consequences for cell growth and potential drug targets.
In addition to detecting disease-relevant mutations, it is therefore crucial to understand the interplay of the affected signaling pathways. Our medical report provides you with a comprehensive overview of tumor-associated signaling networks – including their molecular key players, relevant genetic alterations, and associated drug classes. It helps to understand the complex interactions between signaling pathways and to address potential tumor escape mechanisms specifically. In this way, combination therapies can also be meaningfully evaluated and integrated into therapy planning.
Considered signaling pathways
- Signaling via receptor tyrosine kinases
- Cell cycle
- DNA damage repair
- Hormone pathways
- Wnt pathway
- Hedgehog pathway
- Hippo pathway
- Apoptosis pathway
- Epigenetic regulators
TMB Determination and MSI Prediction
The basis for therapeutic decisions on immunotherapies with checkpoint inhibitors
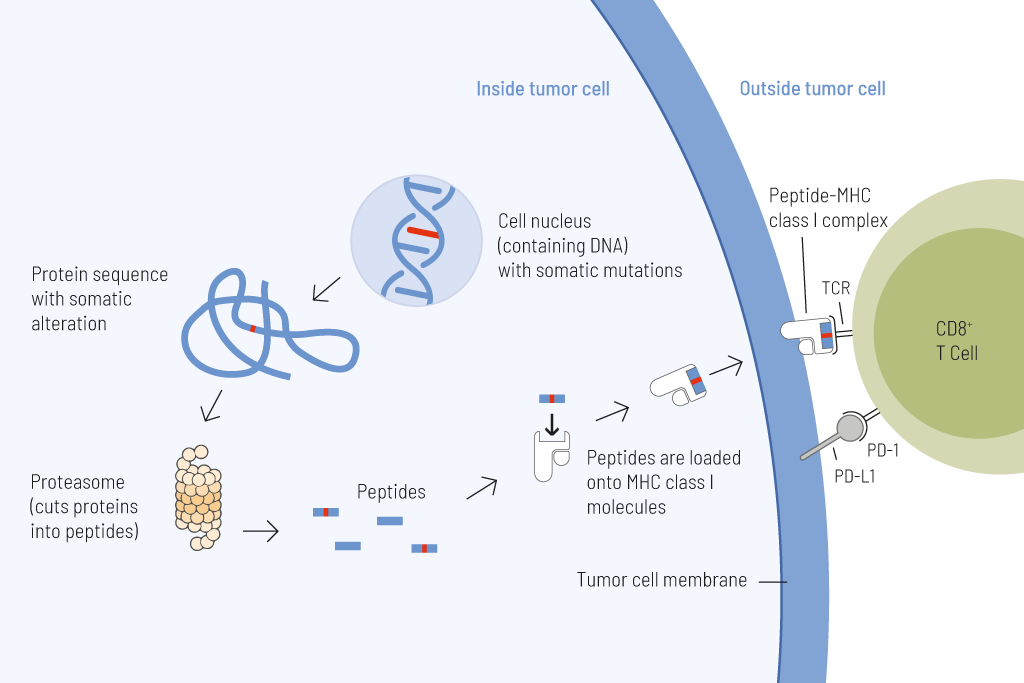
Presentation of tumor cell-derived somatic peptides. Somatic mutations frequently arise in cancer and permanently alter the genomic information. These genetic changes can result in the expression of proteins with altered amino acid sequences. These peptides that carry a somatic change and thus display a particularly strong immunostimulatory potential can be presented on the tumor cell surface and cause an effective anti-tumor immune response.
Tumor mutational burden (TMB), defined as the number of somatic mutations per megabase (Mut/Mb), is an approval-relevant biomarker and predictive of the response to treatment with immune checkpoint inhibitors.
The higher the number of genetic alterations within a tumor cell, the more mutated proteins are formed. These mutated proteins are processed into short fragments (oligopeptides) that are presented on the surface of tumor cells. Such mutated peptides are called neoantigens and are highly immunogenic. This means that they are effectively recognized by immune cells, particularly T cells. Specific T cells are capable of directly eliminating tumor cells after antigen recognition. The higher the number of mutations, the greater the likelihood that neoantigens will be presented on tumor cells, and the more effective tumor elimination by T cells will be. However, T-cell activity can be suppressed by immune checkpoints. Therefore, therapies targeting these checkpoints (so-called immune checkpoint inhibitors) are particularly effective in tumors with high mutational burden.
By comparing tumor and normal tissue, we can calculate the TMB with precision. In our medical report, we provide not only the TMB classification but also the exact mutation value per megabase. This allows for a far more differentiated assessment than the simple categorization into “high” or “low.” Especially in borderline cases, this precise determination enables a more reliable evaluation of whether immunotherapy may be an option.
The accuracy of TMB calculation largely depends on the size of the gene panel analyzed. With 2.96 Mb, CancerPrecision® is well above the recommended minimum size of 1.5 Mb and ensures a robust calculation of mutational burden.
In addition, we determine the MSI status (microsatellite instability), another important marker for immunotherapy response. Microsatellites are short repetitive DNA sequences found throughout the genome. The size of microsatellites can change due to failures in DNA mismatch repair mechanisms when mutations impair the underlying genes. This leads to an accumulation of frameshift mutations, which significantly increase the mutational burden.
Beyond classical PCR methods, prediction in our laboratory is performed fully NGS-based. This enables the analysis of a significantly larger number of microsatellite regions, without requiring a separate laboratory test; the calculation is performed for every sequenced tumor sample. This calculation has been validated with hundreds of matched normal–tumor sample pairs from different tumor types, in which more than 2,500 microsatellite loci were analyzed. Upon request, we also offer the classical PCR-based procedure for MSI determination.
HRD – Homologous Recombination Deficiency
Healthy cells ensure a stable and error-free genome by using different DNA repair mechanisms. Homologous recombination repair (HR) is a DNA repair pathway that acts on DNA double-strand breaks. In case of homologous recombination deficiency (HRD), this pathway is defective so that mutations, chromosomal aberrations, and other errors can accumulate in the genome. Through the resulting genomic instability, HRD facilitates tumor development and has been shown to play a role in various cancers, most prominently in breast and ovarian tumorigenesis5,6.
Loss-of-function genes involved in this pathway can sensitize tumors to PARP inhibitors and platinum-based chemotherapy, which target the destruction of cancer cells by working in concert with HRD through synthetic lethality. To identify tumors where these medications are applicable, reliable determination of the HRD status is of utmost importance.
HR-deficient tumors are often caused by germline or somatic mutations in BRCA1 or BRCA2. Therefore, this pattern has formerly been referred to as BRCAness. Moreover, mutations in other HR genes such as RAD51C, ATM, and PALB2 have been shown to cause HRD. It has to be mentioned that not every genetic defect in HR genes necessarily leads to HRD in the tumor. On the contrary, HRD can be caused without a detectable HR gene mutation, such as promoter methylation of BRCAness genes. Thus, if one tries to detect the mutations only in BRCAness genes, a potential HRD may remain undetected. To ensure that HR-deficient tumors are not overlooked, we calculate the HRD score as part of every CancerPrecision® analysis independent of the tumor entity.
The HRD score measures overall genomic instability based on the number of indels, substitutions and rearrangements occurring on a genome-wide level. The responsible mutations do not have to be precisely identified for this. To calculate the HRD score of the tumor sample, the mutation pattern is used and calculated from three typical HRD events:
- loss of heterozygosity (LOH)
- large-scale state transition (LST)
- telomeric allelic imbalance (TAI)
We report the HRD score in our CancerPrecision® diagnostic report together with any identified somatic mutations and selected gene fusions as well as TMB, MSI and CNVs to provide a most comprehensive tumor analysis.
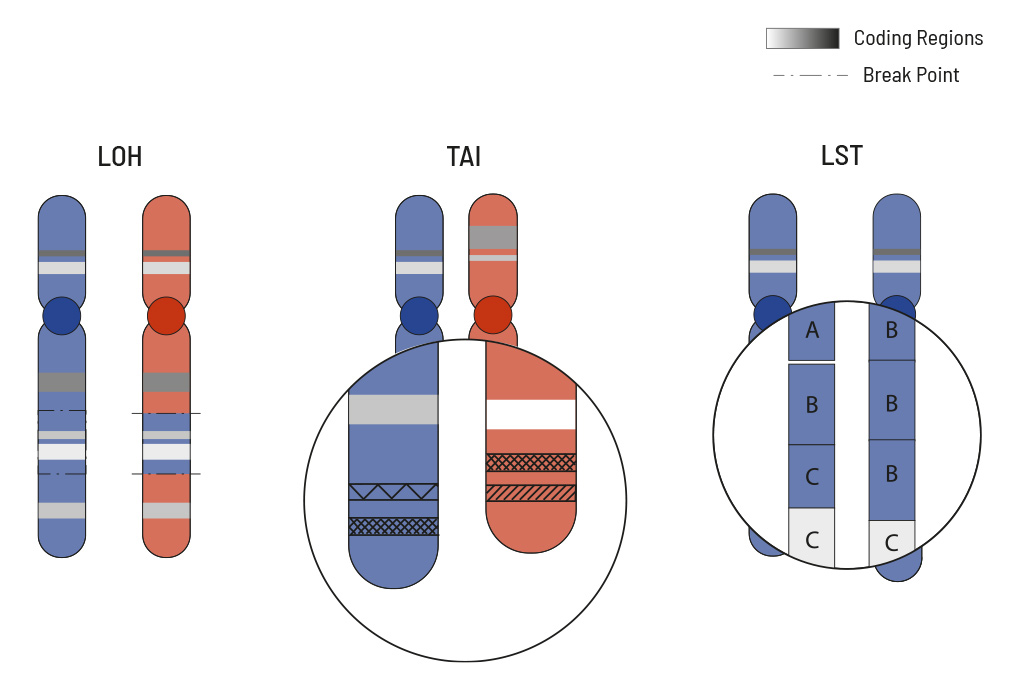
LOH is the irreversible loss of a single parental allele, which is especially severe in cases where defective gene versions are retained. LOH regions are defined as larger than 15 Mb but less than the whole chromosome. TAI occurs when the telomeric end of a chromosome is severely shortened in one of the two parental chromosomes which causes an allelic imbalance in this region. This imbalance occurs because the repetitive DNA sequences in telomere regions are especially sensitive to HRD. LST counts the number of transition points between abnormal chromosome regions that generate chromosomal gains or losses larger than 10 Mb.
CNV Analysis
Determination of deletions/amplifications for highest therapeutic yield
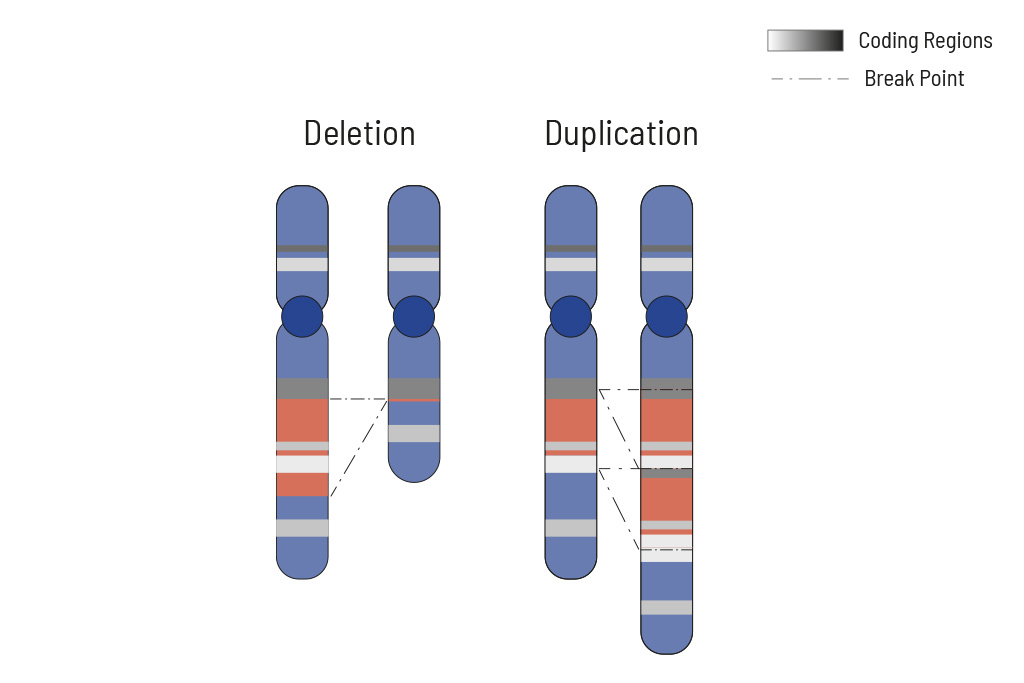
CNVs (copy number variants) often play an important role in tumor genetics. Knowing the changes in CNVs assists in choosing the optimal treatment. Therefore CNV analysis is an integral part of our somatic tumor diagnostics.
Cellular processes are tightly regulated. This regulation depends on the correct function of genes. In tumors, the copy number of genes is frequently altered, thus impairing the affected genes correct function. Increasing the copy number of a gene can increase its activity, while (partial) deletion can result in a loss of function. Therefore, chromosomal aberrations leading to copy number changes can also have therapeutic consequences.
In tumors, CNVs are frequent due to the overall genomic instability. Here, large chromosomal parts are often either deleted or amplified. Understanding these deletions/amplifications and knowing the genes in the affected region with therapeutic relevance is important. Therefore, deletions and amplifications are detected based on the NGS data obtained.
Deletions and amplifications are listed with the affected genes of therapeutic relevance at the beginning of the report. A complete CNV-profile of the analyzed regions is shown in the report’s appendix.
CancerIFP
Multiplex immunofluorescence staining for the analysis of immune and oncoproteins
Conventional immunohistochemistry (IHC) allows the detection of only a single marker per tissue section. In many cases, only limited amounts of tissue are available, restricting the analysis to individual markers. CancerIFP, by contrast, enables the simultaneous analysis of multiple markers on a single tumor tissue section. For each marker, a specific fluorescently labeled antibody is used that selectively binds to its target protein. During image acquisition, fluorescence signals are digitally recorded and separately displayed, providing a precise representation of protein expression and its spatial distribution within the tumor tissue.
CancerIFP provides insights into clinically relevant immune checkpoints and tumor-specific signaling pathways. The analysis includes nine markers, which are examined simultaneously on a single FFPE tissue section. All analyses are performed on our whole-slide immunofluorescence platform and are interpreted by a specialized neuropathologist or pathologist. The analysis was developed in-house as a laboratory-developed test (LDT). Each antibody was internally validated, and results are assessed using a standardized scoring system aligned with established IHC criteria. The results provide a detailed picture of protein expression in tumor tissue and complement our comprehensive tumor diagnostics with protein-level findings.
CancerFusionRx®
RNA-based identification of fusion transcripts
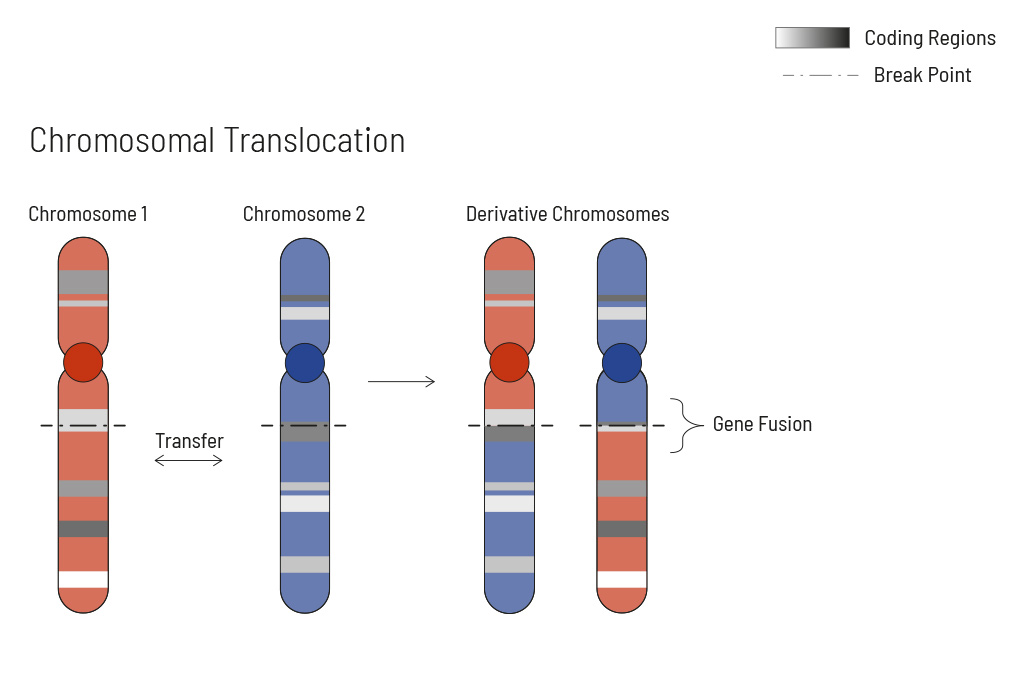
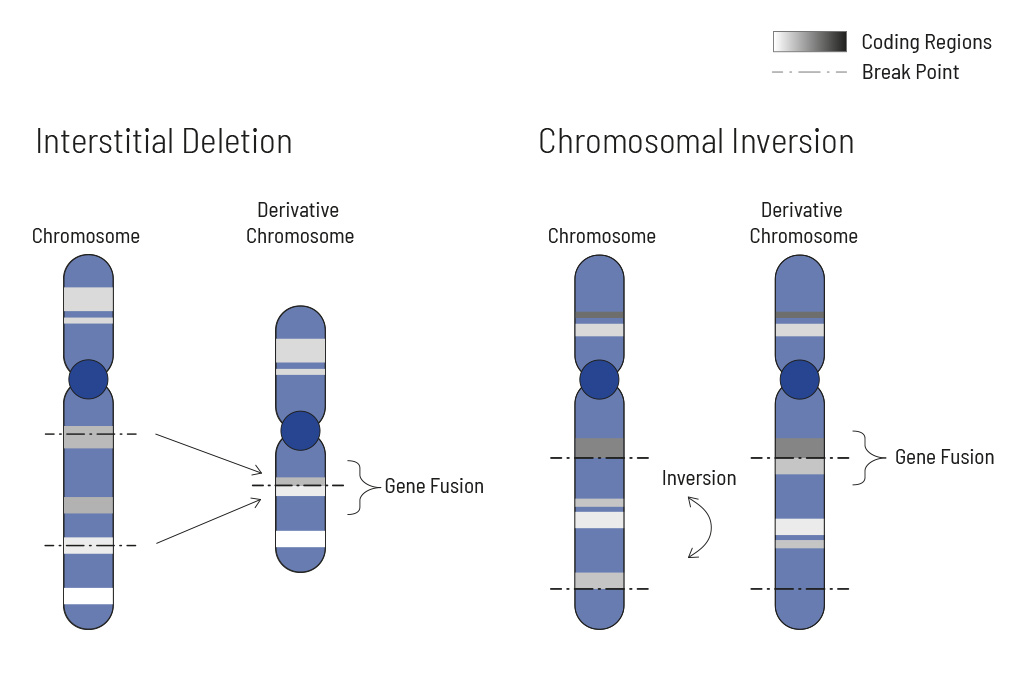
Chromosomal rearrangements frequently occur in all types of cancer. As a result, gene fusions can occur in the cancer genome. Fusions are major drivers of cancer and are therefore most relevant for treatment decisions. Conventional PCR-based methods will not detect a fusion when the other partner is not known (frequently relevant for neutrophic tyrosine kinase, NTRK fusions). Even whole transcriptome analyses are not sensitive enough, especially when the tumor content is low.
To detect all known and previously described as well as novel gene fusions with a therapeutic option, we developed a next-generation targeted enrichment on RNA-basis. The design currently includes over 200 genes for fusion detection and over 132 exon-exon-specific enrichments with known breakpoints. This method is superior to DNA-based methods and also to whole RNA-based approaches. We strongly recommend complementing the genetic tumor diagnostic by RNA enrichment for fusions for the most complete understanding of the tumor’s biology.
Gene Expression Analysis
Tumor RNA analysis for further insights
Gene expression analysis is crucial for improving the understanding of solid tumors at the molecular level. It provides additional information for diagnosis, prognosis, and personalized treatment.
With this add-on service, we perform additional transcriptome sequencing to assess gene expression in all tumor-relevant genes. Aberrant gene expression is determined by comparison with expression patterns from our extensive inhouse cohort of tumor transcriptomes. The results can verify DNA findings on RNA level, provide recommendations for further analysis (such as IHC staining), and identify potential therapeutic targets, even in the absence of detectable DNA mutations.
The findings will be included in the CancerPrecision® report, presented in both table format (covering relevant expression values, such as FPKM per in-house cohort, (Panel A) and violin plots to provide further visual context (Panel B). Possible therapeutic strategies, where applicable, will also be noted in the extensive drug listing in the supplements section.
A)

B)


The X-axis shows the gene expression in FPKM on a logarithmic scale. The red bar marks the level of gene expression of the examined gene of the analyzed sample. The subdivision of the violin plots shown by longitudinal bars represents the percentiles in the respective distribution (percentiles from left to right: 0-25, 25-50, 50-75, and 75-100). Each figure shows from top to bottom: 1) The distribution of gene expression of the indicated gene in a cohort of tumor samples matching the patient’s tumor entity. 2) The distribution of gene expression of the indicated gene in a cohort of tumor samples of other tumor entities. 3) The distribution of gene expression of all expressed genes in the analyzed tumor RNA of the patient.
Gene Directory
Gene list for DNA-based analysis (787 genes, CancerPrecision®, TUM01)
ABCB1, ABCG2, ABL1, ABL2, ABRAXAS1, ACD, ACVR1, ACVR2A, ADGRA2, ADRB1, ADRB2, AIP, AIRE, AJUBA, AKT1, AKT2, AKT3, ALK, ALOX12B, AMER1, ANKRD26, APC, APLNR, APOBEC3A, APOBEC3B, AR, ARAF, ARFRP1, ARHGAP35, ARID1A, ARID1B, ARID2, ARID5B, ASXL1, ASXL2, ATM, ATR, ATRX, AURKA, AURKB, AURKC, AXIN1, AXIN2, AXL, B2M, B4GALNT1, BAP1, BARD1, BAX, BCHE, BCL10, BCL11A, BCL11B, BCL2, BCL2L1, BCL2L11, BCL3, BCL6, BCL9, BCOR, BCORL1, BCR, BIRC2, BIRC3, BIRC5, BLM, BMI1, BMPR1A, BRAF, BRCA1, BRCA2, BRD3, BRD4, BRD7, BRIP1, BTK, BTN3A1, BUB1B, CACNA1S, CALR, CARD11, CASP8, CBFB, CBL, CBLB, CBLC, CCDC6, CCND1, CCND2, CCND3, CCNE1, CD274, CD276, CD70, CD79A, CD79B, CD82, CDC42, CDC73, CDH1, CDH11, CDH2, CDH3, CDH5, CDK1, CDK12, CDK2, CDK4, CDK5, CDK6, CDK8, CDKN1A, CDKN1B, CDKN1C, CDKN2A, CDKN2B, CDKN2C, CEACAM5, CEBPA, CENPA, CEP57, CFTR, CHD1, CHD2, CHD4, CHEK1, CHEK2, CIC, CIITA, CLDN18, CNKSR1, COL1A1, COMT, COQ2, CREB1, CREBBP, CRKL, CRLF2, CRTC1, CSF1R, CSF3R, CSMD1, CSNK1A1, CTAG1B, CTCF, CTLA4, CTNNA1, CTNNB1, CTR9, CTRC, CUL3, CUX1, CXCR4, CYLD, CYP1A2, CYP2A7, CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5, CYP4F2, DAXX, DCC, DDB2, DDR1, DDR2, DDX11, DDX3X, DDX41, DHFR, DICER1, DIS3L2, DLL3, DNMT1, DNMT3A, DOT1L, DPYD, E2F3, EED, EFL1, EGFR, EGLN1, EGLN2, EIF1AX, ELAC2, ELF3, EME1, EML4, EMSY, EP300, EPAS1, EPCAM, EPHA2, EPHA3, EPHB4, EPHB6, ERBB2, ERBB3, ERBB4, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERG, ERRFI1, ESR1, ESR2, ETNK1, ETV1, ETV4, ETV5, ETV6, EWSR1, EXO1, EXT1, EXT2, EZH1, EZH2, EZHIP, F3, FAN1, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FAS, FAT1, FBXO11, FBXW7, FEN1, FES, FGF10, FGF14, FGF19, FGF2, FGF23, FGF3, FGF4, FGF5, FGF6, FGF9, FGFR1, FGFR2, FGFR3, FGFR4, FH, FLCN, FLI1, FLT1, FLT3, FLT4, FOLH1, FOLR1, FOXA1, FOXE1, FOXL2, FOXO1, FOXQ1, FRK, FRS2, FUS, FYN, G6PD, GALNT12, GATA1, GATA2, GATA3, GATA4, GATA6, GGT1, GLI1, GLI2, GLI3, GNA11, GNA13, GNAQ, GNAS, GNB3, GPC3, GPER1, GREM1, GRIN2A, GRM3, GSK3A, GSK3B, GSTP1, H3-3A, H3-3B, H3C1, H3C2, H3C3, HABP2, HAVCR2, HCK, HDAC1, HDAC2, HDAC6, HGF, HIF1A, HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA, HLA-DRB1, HMGA2, HMGCR, HMGN1, HNF1A, HNF1B, HOXB13, HRAS, HSD3B1, HSP90AA1, HSP90AB1, HTR2A, ICOSLG, ID2, ID3, IDH1, IDH2, IDO1, IFNGR1, IFNGR2, IFNL3, IGF1, IGF1R, IGF2, IGF2R, IKBKB, IKBKE, IKZF1, IKZF3, IL1B, IL1RN, IL7R, INPP4A, INPP4B, INPPL1, INSR, IRF1, IRF2, IRS1, IRS2, IRS4, ITPA, JAK1, JAK2, JAK3, JUN, KAT6A, KDM5A, KDM5C, KDM6A, KDR, KEAP1, KIAA1549, KIF1B, KIT, KLF2, KLF4, KLHL6, KLLN, KMT2A, KMT2B, KMT2C, KMT2D, KRAS, KSR1, LAG3, LAMP1, LATS1, LATS2, LCK, LIG4, LIMK2, LRP1B, LRRK2, LTK, LYN, LZTR1, MAD2L2, MAF, MAGEA1, MAGEA12, MAGEA3, MAGEA4, MAGEA8, MAGI1, MAGI2, MAML1, MAP2K1, MAP2K2, MAP2K3, MAP2K4, MAP2K5, MAP2K6, MAP2K7, MAP3K1, MAP3K13, MAP3K14, MAP3K3, MAP3K4, MAP3K6, MAP3K8, MAPK1, MAPK11, MAPK12, MAPK14, MAPK3, MAX, MBD4, MC1R, MCL1, MDC1, MDH2, MDM2, MDM4, MECOM, MED12, MEF2B, MEN1, MERTK, MET, MGA, MGMT, MITF, MLH1, MLH3, MLLT10, MLLT3, MMP2, MMS22L, MN1, MPL, MRE11, MS4A1, MSH2, MSH3, MSH4, MSH5, MSH6, MSLN, MSR1, MST1R, MTAP, MTHFR, MTOR, MT-RNR1, MTRR, MUC1, MUTYH, MXI1, MYB, MYC, MYCL, MYCN, MYD88, MYH11, MYH9, MYOD1, NAT2, NBN, NCOA1, NCOA3, NCOR1, NF1, NF2, NFE2L2, NFKB1, NFKB2, NFKBIA, NFKBIE, NIN, NKX2-1, NLRC5, NOTCH1, NOTCH2, NOTCH3, NOTCH4, NPM1, NQO1, NR1I3, NRAS, NRG1, NSD1, NSD2, NSD3, NT5C2, NTHL1, NTRK1, NTRK2, NTRK3, NUDT15, NUMA1, NUP98, NUTM1, OBSCN, OPRM1, PAK1, PAK3, PAK4, PAK5, PALB2, PALLD, PARP1, PARP2, PARP4, PAX3, PAX5, PAX7, PBK, PBRM1, PBX1, PDCD1, PDCD1LG2, PDGFA, PDGFB, PDGFC, PDGFD, PDGFRA, PDGFRB, PDK1, PDPK1, PGR, PHF6, PHOX2B, PIAS4, PIGA, PIK3C2A, PIK3C2B, PIK3C2G, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PIK3R1, PIK3R2, PIK3R3, PIM1, PLCG1, PLCG2, PLK1, PMEL, PML, PMS1, PMS2, POLB, POLD1, POLE, POLH, POLQ, POR, POT1, PPARG, PPM1D, PPP2R1A, PPP2R2A, PRAME, PREX2, PRKAR1A, PRKCA, PRKCI, PRKDC, PRKN, PRMT5, PRR4, PSMB1, PSMB10, PSMB2, PSMB5, PSMB8, PSMB9, PSMC3IP, PSME1, PSME2, PSME3, PTCH1, PTCH2, PTEN, PTGS2, PTK2, PTK7, PTPN11, PTPN12, PTPRC, PTPRD, PTPRS, PTPRT, RABL3, RAC1, RAC2, RAD21, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD54B, RAD54L, RAF1, RALGDS, RARA, RASA1, RASAL1, RB1, RBM10, RECQL4, REST, RET, RFWD3, RFX5, RFXANK, RFXAP, RHBDF2, RHEB, RHOA, RICTOR, RIF1, RINT1, RIPK1, RIT1, RNASEL, RNF43, ROS1, RPS20, RPS6KB1, RPS6KB2, RPTOR, RSF1, RSPO1, RSPO2, RSPO3, RSPO4, RUNX1, RYR1, SAMHD1, SAV1, SBDS, SCG5, SDHA, SDHAF2, SDHB, SDHC, SDHD, SEC23B, SERPINB9, SETBP1, SETD2, SETDB1, SF3B1, SGK1, SH2B3, SHH, SHLD2, SIK2, SKP2, SLC19A1, SLC26A3, SLC45A2, SLCO1B1, SLFN11, SLIT2, SLX4, SMAD3, SMAD4, SMARCA2, SMARCA4, SMARCB1, SMARCE1, SMC1A, SMC3, SMO, SOCS1, SOS1, SOX11, SOX2, SOX9, SPEN, SPINK1, SPOP, SPRED1, SRC, SRD5A2, SRGAP1, SRSF2, SSTR2, SSX1, STAG2, STAT1, STAT3, STAT5A, STAT5B, STK11, SUCLG2, SUFU, SUZ12, SYK, TACSTD2, TAF1, TAF15, TAP1, TAP2, TAPBP, TBK1, TBX3, TCF3, TCF4, TCL1A, TEK, TERC, TERF2IP, TERT, TET1, TET2, TFE3, TGFB1, TGFBR2, TMEM127, TMPRSS2, TNFAIP3, TNFRSF13B, TNFRSF14, TNFRSF8, TNFSF11, TOP1, TOP2A, TP53, TP53BP1, TP63, TPMT, TPX2, TRAF2, TRAF3, TRAF5, TRAF7, TRIM28, TRRAP, TSC1, TSC2, TSHR, TTK, TYMS, U2AF1, UBE2T, UBR5, UGT1A1, UGT2B15, UGT2B7, UIMC1, USP9X, VEGFA, VEGFB, VHL, VKORC1, VTCN1, WRN, WT1, XIAP, XPA, XPC, XPO1, XRCC1, XRCC2, XRCC3, XRCC5, XRCC6, YAP1, YES1, ZFHX3, ZNF217, ZNF703, ZNRF3, ZRSR2
DNA-based detection of selected structural variations in these genes
ALK, BCL2, BCOR, BCR, BRAF, BRD4, CDKN2A, CDKN2B, EGFR, ERG, ETV4, ETV6, EWSR1, FGFR1, FGFR2, FGFR3, FUS, MET, MSH2, MYB, MYC, NFE2L2, NOTCH2, NRG1, NTRK1, NTRK2, NTRK3, PAX3, PDGFB, RAF1, RARA, RET, ROS1, SSX1, SUZ12, TAF15, TCF3, TFE3, TMPRSS2
Gene list for RNA-based identification of fusion transcripts (CancerFusionRx®, STR01)
Gene list for de-novo fusion detection
ABL1, ACTB, AFAP1, AGK, AKAP4, AKAP9, AKAP12, AKT1, AKT2, AKT3, ALK, ARHGAP6, ARHGAP26, ASPL, ASPSCR1, ATF1, ATP1B1, ATRX, AVIL, AXL, BAG4, BCL2, BCOR, BCORL1, BCR, BEND2, BICC1, BRAF, BRD3, BRD4, c11orf95, CAMTA1, CCAR2, CCDC6, CCDC88A, CCDC170, CCNB3, CCND1, CD44, CD74, CEP85L, CIC, CLDN18, CLIP1, CLTC, CNTRL, COL1A1, CREB1, CREB3L1, CREB3L2, CRTC1, CTNNB1, DDIT3, DNAJB1, EGFR, EML4, EPC1, EPCAM, ERBB2, ERBB4, ERG, ESR1, ESRRA, ETV1, ETV4, ETV5, ETV6, EWSR1, EZR, FEV, FGFR1, FGFR2, FGFR3, FLI1, FN1, FOXO1, FOXO4, FOXR2, FUS, GLI1, GOPC, GPR128, HEY1, HMGA2, HTRA1, IGF1R, INSR, JAK2, JAZF1, KIAA1549, KIF5B, KIT, LEUTX, LMNA, LPP, LTK, MAGI3, MAML1, MAML2, MAML3, MAMLD1, MAP3K8, MARS1, MAST1, MAST2, MEAF6, MET, MGA, MGMT, MITF, MKL2, MN1, MSH2, MYB, MYBL1, MYC, NAB2, NCOA1, NCOA2, NCOA3, NCOA4, NFATC2, NFIB, NOTCH2, NPM1, NR4A3, NRG1, NRG2, NSD3, NTRK1, NTRK2, NTRK3, NUTM1, PAX3, PAX7, PAX8, PBX1, PDGFB, PDGFD, PDGFRA, PDGFRB, PHF1, PIK3CA, PLAG1, PML, POU5F1, PPARG, PPARGC1A, PPP1CB, PRKACA, PRKAR1A, PRKCA, PRKCB, PRKD1, PRKD2, PRKD3, PTPRZ1, QKI, RAD51B, RAF1, RANBP2, RARA, RELA, RELCH, RET, ROS1, RPS6KB1, RREB1, RSPO2, RSPO3, SDC1, SDC4, SH3PXD2A,SLC1A2, SHTN1, SLC34A2, SLC44A1, SLC45A3, SND1, SQSTM1, SS18, SSX1, SSX2, SSX4, STAT6, STRN, SUZ12, TACC1, TACC2, TACC3, TAF2N, TAF15, TCF3, TCF12, TERT, TFE3, TFEB, TFG, THADA, TMPRSS2, TPM3, TPR, TRIM24, TRIM33, TRIO, TTYH1, VGLL2, VGLL3, VMP1, WT1, WWTR1, YAP1, YWHAE, ZC3H7B, ZMYM2, ZNF703
Gene list for selected break points in these fusion genes
AFAP1-NTRK2, ATP1B1-NRG1, BCOR-CCNB3, BRD3-NUTM1, BRD4-NUTM1, CCDC6–RET, CCDC88A-ALK, CD74-NRG1, CD74-ROS1, CLTC-ALK, DNAJB1-PRKACA, EGFR-PPARGC1A, EML4-ALK, ETV6 NTRK2, ETV6-NTRK3, EWSR1-ATF1, EWSR1-ERG, EWSR1-FLI1, EWSR1-WT1, EZR-ROS1, FGFR1-TACC1, FGFR2-BICC1, FGFR2 TACC3, FGFR3-TACC3, KIAA1549-BRAF, KIF5B-ALK, KIF5B–RET, MGA-NUTM1, NAB2-STAT6, NCOA4–RET, NPM1-ALK, NSD3-NUTM1, PAX3-FOXO1, PAX7-FOXO1, PPP1CB-ALK, PRKAR1A-RET, QKI-NTRK2, RANBP2- ALK, RPS6KB1–VMP1, SDC4-NRG1, SDC4 ROS1, SLC34A2-ROS1, SND1-BRAF, SS18-SSX1, SS18-SSX2, STRN ALK, TMPRSS2-ERG, TPM3-ALK, TPM3-NTRK1, TPM3-ROS1, TPR NTRK1, TRIM24-BRAF, TRIM24-NTRK2, TRIM33-RET, TRIO-TERT
List for specific transcript variants
EGFR del ex2-22 (mLEEK), EGFR del ex25-26 (EGFRvIVb), EGFR del ex25-27 (EGFRvIVa), EGFR del ex26-27, EGFR del ex14-15 (vII), EGFR del ex2-7 (vIII), FGFR2IIIb, MET ex14 skipping, NFE2L2 ex2 skipping, PDGFRA del ex8-9
References
1 Jones, S. et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Science translational medicine 7, 283ra53; 10.1126/scitranslmed.aaa7161. (2015).
2 Sun, J. X. et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS computational biology 14, e1005965; 10.1371/journal.pcbi.1005965 (2018).
3 Nassar, A. H. et al. Ancestry-driven recalibration of tumor mutational burden and disparate clinical outcomes in response to immune checkpoint inhibitors. Cancer cell 40, 1161-1172.e5; 10.1016/j.ccell.2022.08.022 (2022).
4 Buchhalter, I. et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. International journal of cancer 144, 848–858; 10.1002/ijc.31878 (2019).
5 Heeke, A. L. et al. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO precision oncology 2018; 10.1200/PO.17.00286 (2018).
6 Nguyen, L., W M Martens, J., van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nature communications 11, 5584; 10.1038/s41467-020-19406-4 (2020).
Further Information
Webinar: Discover the Power of Modern Tumor Diagnostics
Genetic tumor diagnostics can save lives
Downloads
Contact Us
Do you have a question, or are you interested in our service?
Diagnostic Support
We will assist you in selecting the diagnostic strategy – for each patient.
