The genome represents the entire genetic information of an organism. The most comprehensive collection of an individual’s genetic information is provided by analyzing the whole genome using next-generation sequencing (NGS) technology. By comparing the individual’s genetic information to a reference genome single nucleotide variants (SNVs), small insertions and deletions (indels), copy number variations (CNVs), and structural variants (SVs) can be studied.
Whole genome sequencing studies contribute to:
- cancer studies, personalized medicine approaches, and translational research
- discovery of biomarkers and understanding of pharmacogenetics
- disease research
- plant and animal breeding programs
- examination of microorganisms
You can choose between various Whole Genome Sequencing products to receive the comprehensive genome analysis you need for your research project.


CeGaT Is the Best Partner for Sequencing Your Project
Our Commitment to You
Fast Processing
Turnaround time
≤ 15 business days
High Quality
Highest accuracy for all processes
Secure Delivery
Secure provision of sequenced data via in-house servers
Safe Storage
Safe storage of samples and data after project completion
Our Service
We provide a comprehensive and first-class project support – from selecting the appropriate product to evaluating the data. Each project is supervised by a committed scientist. You will have a contact person throughout the whole project.
Our service includes:
- detailed project consulting
- product selection tailored to your project
- detailed bioinformatic evaluation of your data
- detailed project report with information about sample quality, sequencing parameters, bioinformatic analysis, and results
Benefit from our dedicated support and accredited workflows.

Explore Our Product Portfolio for Genome Sequencing
We offer different Whole Genome Sequencing (WGS) products to address a variety of research questions. Would you like to have bioinformatic analyses performed on your data in addition to the included deliverables? Each of our products can be supplemented with further services. We are happy to advise you.
WGS Large Classic | WGS Small Classic | WGS Flex | HiFi WGS |
Species | Species | Species | Species |
DNA quality | DNA quality | DNA quality | DNA quality |
Protocol +/- PCR amplification | Protocol + PCR amplification | Protocol +/- PCR amplification | Protocol - PCR amplification |
Read length | Read length | Read length | Read length |
Sequencing platform | Sequencing platform | Sequencing platform | Sequencing platform |
Output | Output | Output | Output |
Included deliverables Project report & files in FASTQ format | Included deliverables | Included deliverables | Included deliverables |
Bioinformatics
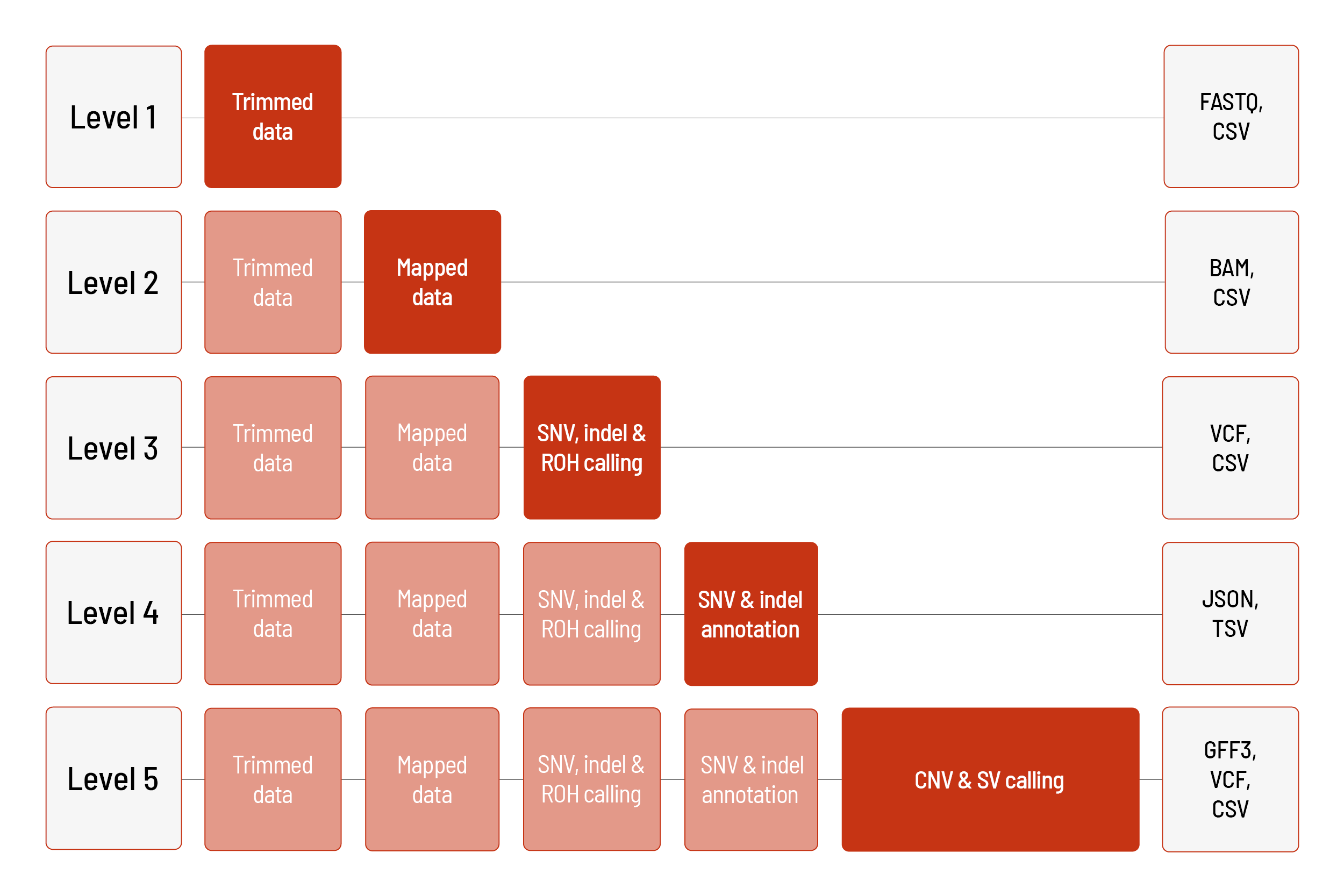
Raw sequencing data are automatically processed. We offer different levels of bioinformatic analysis. The default level is Level 1. With increasing bioinformatic level, more data are delivered. All higher levels include the data from the lower levels. In addition to the data, and independent of the analysis level, a project report is generated.
Analysis of DNA Samples with the Illumina Platform For:
- WGS Large Classic
- WGS Small Classic
- WGS Flex
WGS Large Classic and WGS Flex are analyzed with the Illumina DRAGEN Bio-IT Platform. WGS Small Classic is analyzed with the CeGaT pipeline.
For human samples, we can perform the analyses based on the human references hg19 or GRCh38.
Level 1:
- demultiplexing and adapter trimming of the sequencing data (FASTQ format)
- metrices (CSV format)
- MultiQC report (HTML format)
Level 2:
- mapping of the sequencing data (BAM format)
- prediction of ploidy of sex chromosomes (CSV file) (only for human samples)
- metrices (CSV format)
Level 3:
- calling of single nucleotide variants (SNVs) and small insertions and deletions (indels) (VCF format)
- calling of regions of homozygosity (ROH) (BED and CSV format) (only for human samples)
- metrices (CSV format)
Level 4:
- annotation of the SNVs and indels (JSON and TSV format) (only for human samples)
Level 5:
- calling of copy number variations (CNVs) (VCF and GFF3 format) including annotations (JSON and TSV format) (annotations only for human samples)
- detection of structural variants (SVs), such as translocations, inversions as well as large and medium-sized indels (VCF and CSV format) including annotations (JSON and TSV format) (annotations only for human samples)
- metrices (CSV format)
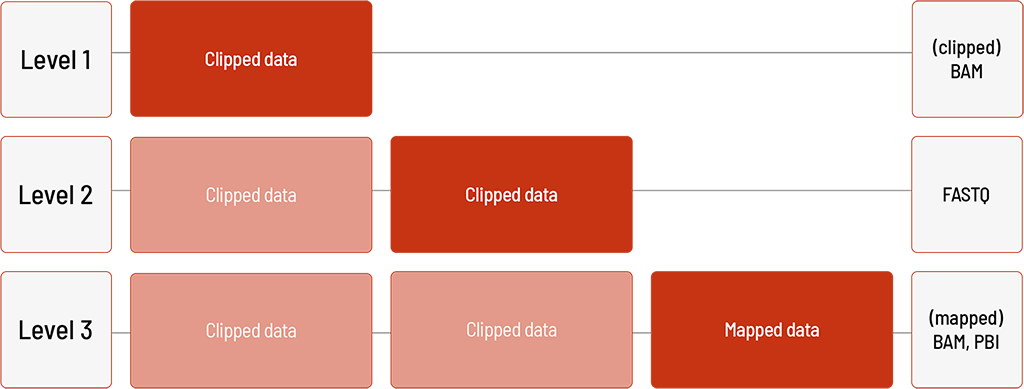
Analysis of DNA Samples with the PacBio Platform For:
- HiFi WGS
Our HiFi WGS product is analyzed with the PacBio Platform.
For human samples, we can perform the analyses based on the human references hg19 or GRCh38. Level 3 and all higher levels are only available for human samples.
Level 1:
- clipped sequencing data in FASTQ format
- if required, clipped sequencing data in BAM format can additionally be delivered
Level 2:
- mapping of the sequencing data (BAM and BAI format)
Level 3:
- calling of single nucleotide variants (SNVs) (VCF format)
Level 4:
- annotation of the SNVs (JSON and TSV format)
Level 5:
- detection of structural variants (SVs) (VCF format) including annotations (JSON and TSV format)
Technical Information
At CeGaT, we are using sequencing technologies from Illumina and Pacific Bioscience (PacBio). For the products using the Illumina sequencing platforms, WGS samples are sequenced in 2 x 150 bp or 2 x 100 bp mode. Our PacBio platform enables us to sequence DNA fragments of up to 18 kb. If you require other sequencing parameters, please let us know! We can provide further solutions.
Further Information about Genome Sequencing
Genome sequencing is also known as whole genome sequencing (WGS), full genome sequencing, or complete genome sequencing. Using this approach, the entirety of the genomic information is sequenced. Thus, the complete genome of a sample is analyzed at a single time. This means that in addition to the chromosomal DNA, the mitochondrial and, if available, the chloroplast’s DNA is sequenced. Whole genome sequencing is, thus, available for a variety of different organisms, including microorganisms, viruses, plants, and mammals, such as humans or mice.
With the development of high-throughput sequencing, formerly known as next-generation sequencing, the costs for whole genome sequencing dropped drastically, making research with whole genome sequencing possible and affordable. Whole genome sequencing has a variety of application areas. It can, for example, be used, to determine the mutation frequency in the complete human genome. Mutations occur naturally, e.g., due to replication errors during cell divisions, or DNA damaging and subsequent error-prone or insufficient repair mechanisms.
The mutation frequency is furthermore increased in cancer due to genomic instability. Altered mutation frequencies can be observed using whole genome sequencing. Another application area for whole genome sequencing includes genome-wide association studies, in which genetic variants or variants associated with a phenotype, e.g., a disease, are researched.
Whole genome sequencing is not only a powerful tool in the scientific field, it has also diagnostic applications. It can, for example, be used for diagnosing and treating diseases, including rare diseases, or detecting pathogens, and tracking outbreaks. With whole genome sequencing, personalized medicine is attainable and affordable to improve public health. In contrast to whole exome sequencing, whole genome sequencing also includes the intronic regions of a genome, and, thus, covers all coding and non-coding regions of the genome. This complete sequencing of the genome allows its more comprehensive assessment.
With whole genome sequencing getting faster, easier, and less expensive, it can contribute to your research today!
Downloads
Contact Us
Do you have a question or are you interested in our service? Feel free to contact us. We will take care of your request as soon as possible.
Start Your Project with Us
We are happy to discuss sequencing options and to find a solution specifically tailored to your clinical study or research project.
When getting in contact, please specify sample information including starting material, number of samples, preferred library preparation option, preferred sequencing depth and required bioinformatic analysis level, if possible.
