The personalization of cancer treatment is evolving rapidly. Developing a personalized cancer vaccine is a promising strategy for cancer treatment.
Body cells constantly present peptides on their surface, indicating the current state of the cell. Immune cells can recognize if a cell presents normal or abnormal peptides and use this information to determine and eliminate defective cells. Abnormal peptides, so-called neoantigens, arise from DNA mutations in tumor cells and are targets for therapy using personalized vaccines.
CeGaT’s CancerNeo® enables the analysis of a patient’s tumor exome to detect tumor-specific (somatic) mutations, identifies the HLA-types, and predicts neoantigens. The expression of these neoantigens is confirmed by whole RNA sequencing (transcriptome) from the same tumor sample. Thus, CancerNeo® provides the insights required for the design of personalized cancer vaccines – a powerful tool to boost the immune system’s response to cancer cells.

What We Offer with This Service
Our Promise to You

Service Details
- Whole exome sequencing of tumor/normal tissue using CeGaT’S ExomeXtra®
- Detailed assessment of somatic variants detected in more than 700 tumor-relevant genes and fusions in more than 30 genes
- Medical report with:
- validated list of variants with potential therapeutic relevance
- treatment options based on somatic variants
- TMB determination/MSI prediction/ HRD score calculation/ detection of viral infections (HPV, EBV, MCV, CMV)
- detection of copy number variants (CNVs)
- a list of all eligible drugs, with EMA and/or FDA approval, for which corresponding biomarkers could be detected in the tumor
- determination of pharmacogenetically relevant germline variants affecting the metabolism of certain tumor drugs or anesthetics
- assessment of the evidence for CHIP (Clonal Hematopoiesis of Indeterminate Potential)
- Transcriptome sequencing for gene expression analysis, listing relevant findings based on aberrant gene expression by comparison case expression values with expression patterns from our large inhouse cohort
- Tumor whole RNA sequencing with rRNA depletion
- HLA typing
- Prediction of HLA class I restricted peptide epitopes (neoepitopes) spanning tumor-specific variants from sequencing data
- Selection of most relevant neoepitopes for HLA class I and HLA class II
- Second medical report with selected peptides for formulation of a vaccine
Optional Services
- RNA-based fusion transcript analysis (CancerFusionRx®) covering over 150 genes for fusion detection and over 120 exon-exon-specific enrichments with known breakpoints — learn more
- Immunohistochemical (IHC) analyses: PD-L1, CAR-T cell panel, HLA class I and class II staining (external) — learn more
- MGMT promoter methylation analysis
Sample Report
Our Standard Sample Requirements
Normal Tissue
- 1–2 ml EDTA blood (recommended sample type) or
- Genomic DNA (1–2 µg)
Tumor Tissue
Tumor content at least 20%
- FFPE tumor block (min. tissue size 5x5x5 mm) (recommended sample type) or
- FFPE tumor tissue slides (min. 10 slices 4-10 µm, tissue size 5×5 mm) or
- Genomic DNA (> 200 ng) or
- Fresh frozen tumor tissue or
- 3x 10 ml cfDNA tubes for liquid biopsy
Here you can find more information on how to ship your sample safely.
Further Sample Materials
Other sample material sources are possible on request. Please note: in case of insufficient sample quality or tumor content, the analysis might fail.
If you have more than one option of tumor samples, please get in touch with us (tumor@cegat.com), and we will assist you in choosing the optimal specimen for your patient.
For highest accuracy, we require tumor and normal tissue for our somatic tumor diagnostics panel.
This Is What Makes Our CancerNeo® Service Special
Developing a personalized cancer vaccine requires the highest precision in identifying somatic variants, as well as in the subsequent prediction and selection of neoantigens for vaccine design. CeGaT’s CancerNeo® identifies somatic alterations through comparative analysis of tumor and normal tissue. The analysis is based on CeGaT’S ExomeXtra®, the best possible exome enrichment, and is the most accurate method for identifying somatic variants. In addition, transcriptome sequencing identifies highly expressed potential targets for vaccine design and is part of our selection process based on our long experience. Thus, CancerNeo® reliably identifies the most promising neoantigens.
Additional Panel Sequencing
The basis of analysis for CancerNeo® is whole-exome sequencing, as it is necessary to understand all somatic mutations for the prediction of neoantigens. Our specialized somatic tumor panel enrichment, the basis for our CancerPrecision® diagnostic service, focused the analysis on tumor-associated genes and selected translocations. These are therefore sequenced in much higher resolution. The additional panel sequencing option bases the medical report for treatment decision support (CancerPrecision®) on the sequencing data from the specialized panel enrichment while the medical report for predicted neoantigens usable for a personalized vaccination approach is based on the whole exome sequencing data. The CancerPrecision® report benefits from the higher sequencing resolution and the possibility of detecting the selected translocations enriched by the panel sequencing.
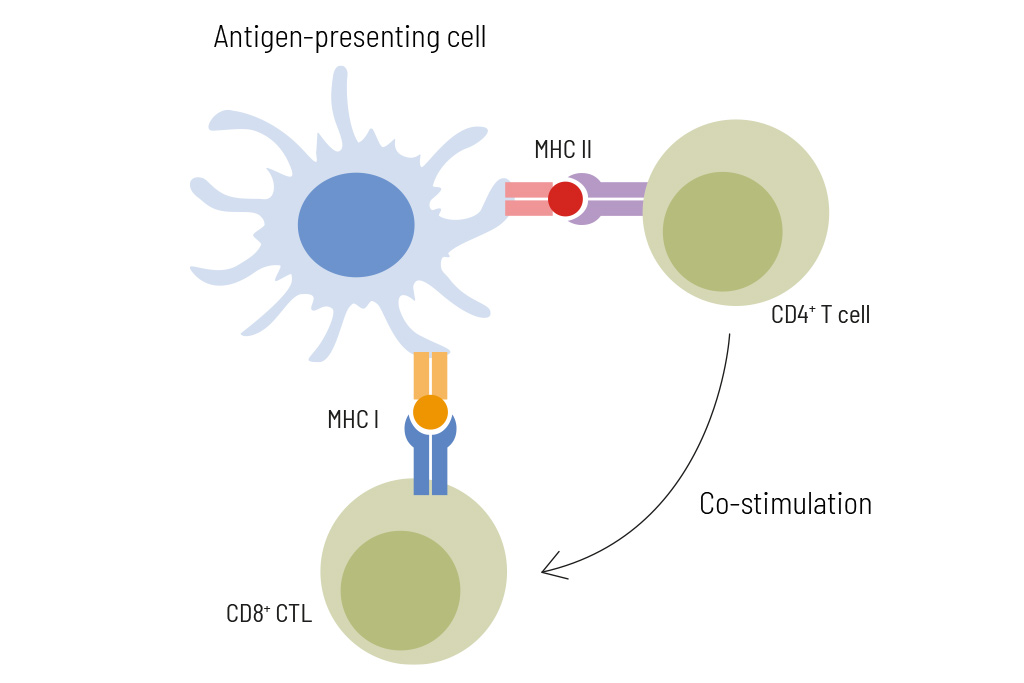
What Is a Neoantigen Prediction?
Neoepitopes can be recognized by CD8+ cytotoxic T cells (CTL) and by CD4+ T helper cells. Neoepitopes recognized by CD8+ T cells are typically 8-12 amino acids long and presented by HLA class I molecules, while neoepitopes recognized by CD4+ T helper cells are usually longer (13-18 amino acids in length) and presented by HLA class II molecules. For successful immunotherapy, neoantigen selection should include short and long peptides to activate both, CD4+ and CD8+ T cells. Immune responses of each individual patient can be monitored during anti-cancer vaccination by flow cytometry-based analysis of neoepitope-specific T cell activation.
Our cancer immunology experts use in silico algorithms based on exome sequencing and HLA typing to predict and select up to 12 eligible neoantigen epitopes individualized for each patient. The selected peptides are predicted to activate not only cytotoxic T cells but also T helper cells. Therefore, in addition to short peptides (8-12 amino acids) potentially binding to HLA class I molecules also long peptides (~17 amino acids) potentially binding to HLA class II molecules are included. To confirm the expression of the selected neoepitopes, transcriptome data by RNA sequencing of the tumor sample is done in parallel. In cases of no availability of RNA, information about neoepitope expression is retrieved from protein expression databases
Immunohistochemistry (IHC) Analyses
To complete our genetic diagnostics, we offer to organize immunohistochemistry analysis on the tumor sample in cooperation with partner laboratories. We forward the pathological examination reports upon completion.
PD-L1
Detection of expression in the tumor tissue of the Programmed death-ligand 1 (PD-L1) is important for selecting patients which may benefit (the most) from immunotherapy, such as pembrolizumab.
MGMT promotor methylation analysis
Detection of MGMT promotor methylation in the tumor tissue is important for glioma patients since it is a potential biomarker of sensitivity to alkylating chemotherapy, including temozolomide (TMZ).
HLA Class I and class II
HLA molecules present tumor antigens to T cells. Expression analysis of HLA-molecules is a helpful tool to tailor your immune-therapeutic strategy.
CAR T cell panel (GD2, EGFR, IL13Ralpha, CD276, HER2, PSMA, ROR1, CD47)
The CAR T cell panel detects eight different target antigens of CAR-T Cell therapies in clinical or preclinical development.
CancerFusionRx®
RNA-based identification of fusion transcripts
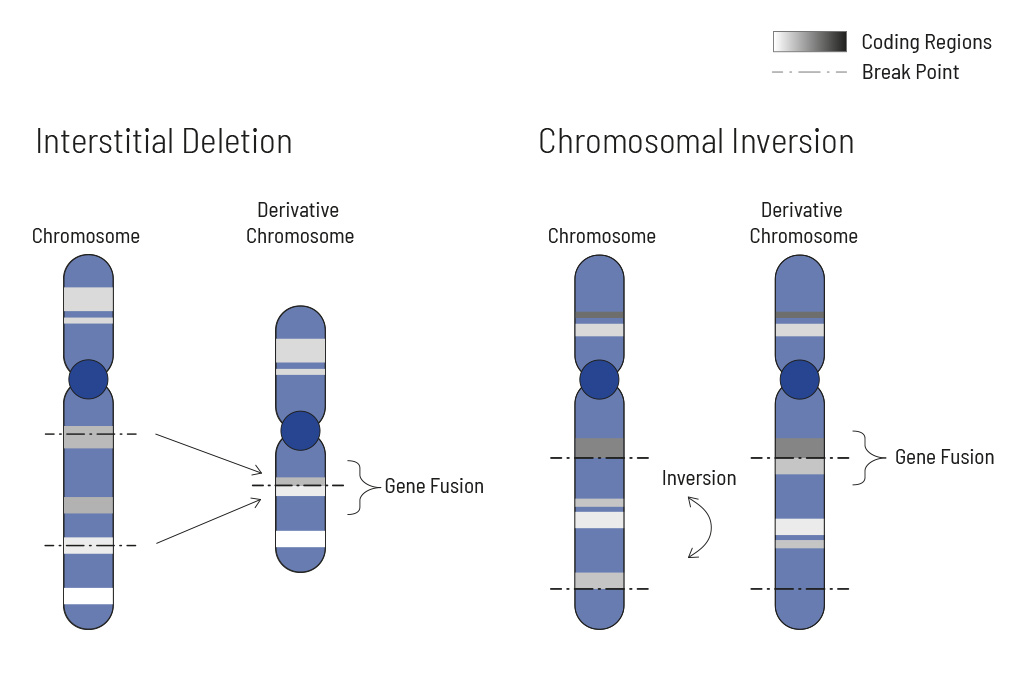
Chromosomal rearrangements frequently occur in all types of cancer. As a result, gene fusions can occur in the cancer genome. Fusions are major drivers of cancer and are therefore most relevant for treatment decisions. Conventional PCR-based methods will not detect a fusion when the other partner is not known (frequently relevant for neutrophic tyrosine kinase, NTRK fusions). Even whole transcriptome analyses are not sensitive enough, especially when the tumor content is low.
To detect all known and previously described as well as novel gene fusions with a therapeutic option, we developed a next-generation targeted enrichment on RNA-basis. The design currently includes over 200 genes for fusion detection and over 132 exon-exon-specific enrichments with known breakpoints. This method is superior to DNA-based methods and also to whole RNA-based approaches. We strongly recommend completing the genetic tumor diagnostic by RNA enrichment for fusions for the most complete understanding of the tumor’s biology.
Contact Us
Do you have a question, or are you interested in our service?
Diagnostic Support
We will assist you in selecting the diagnostic strategy – for each patient.
